Дефект межпредсердной перегородки
Содержание:
Симптомы стеноза почечной артерии
В большинстве случаев стеноз может явно не проявляться, особенно в начальной стадии развития. В этот период больной может жаловаться на периодические головные боли, легкую слабость, эмоциональную неустойчивость. При обследовании области живота у пациента могут прослушиваться несильные шумы по всей верхней области. В некоторых случаях наблюдается рост диастолического давления.
Ключевым симптомом недуга считается артериальная гипертензия, которая очень сложно поддается терапии. В данном случае у человека может наблюдаться ишемическая сердечная болезнь или перемежающаяся хромота.
При продолжительном течении болезни самочувствие человека продолжает ухудшаться.
Существует целый ряд признаков, при выявлении которых необходимо обращаться к врачу:
- ранняя форма гипертонии;
- высокое давление;
- увеличение количества симптомов и их усиление их проявлений;
- неконтролируемые и неподдающиеся регулированию с помощью медикаментов суточные колебания давления, например, повышение показателей в ночное время;
- дисфункция почек, которая проявляется уменьшением скорости клубочковой фильтрации и ростом креатинина в крови;
- тяжелые осложнения в виде дисфункции кровообращения мозга, сердечной недостаточности.
При таких симптомах прием ингибиторов АПФ и блокаторов ангиотензиновых рецепторов не только не принесет должного эффекта, а только ухудшает работу органа. Почечная недостаточность также увеличивается от приема нестероидных средств противовоспалительного назначения и диуретиков.
Дисфункцию почек провоцирует также холестериновая эмболия, возникающая от полученных травм или употребления антикоагулянтов в больших дозировках. В данном случае наблюдаются такие симптомы:
- боли в пояснице;
- олиго- или анурия;
- трансформация мочевого осадка;
- рост креатинина в крови;
- гиперкалиемия.
Помимо вышеперечисленных проявлений страдают другие органы и системы, а именно:
- мозговые артерии – приступы головной боли, тошноты, рвота, ишемические атаки, инсульт;
- глазная сетчатка – отечность зрительного нерва, нарушение кровообращения;
- сосуды органов пищеварения – запоры, внутренние кровотечения, непроходимость, ишемического генеза;
- сосуды кожи.
С возрастом вышеперечисленные негативные проявления ухудшаются и могут совмещаться с другими заболеваниями, провоцируя нефропатию, пиелонефрит, мочекаменную болезнь, поражение почек вследствие приема медикаментов.
Аортальный стеноз
Что такое аортальный стеноз?
Аортальный стеноз — это сужение аорты, самого крупного сосуда в нашем организме, который выполняет функцию транспортных путей и несет насыщенную кислородом кровь от сердца ко всем нашим органам и тканям.
Это сужение может встречаться на разных уровнях, и в зависимости от места сужения, различают следующие виды аортального стеноза: клапанный (сужение клапана аорты), надклапанный (собственно самой аорты), подклапанный (сужение, обусловленное чрезмерным разрастанием мышц левого желудочка, препятствующее выходу крови в аорту).
Наиболее часто встречается клапанный стеноз аорты (Рис 1). В норме аортальный клапан позволяет насыщенной кислородом (артериальной) крови свободно поступать из левого желудочка (насос) в аорту, которая несет кровь ко всем органам и тканям нашего организма. Клапан аорты состоит из трех створок. Когда левый желудочек сокращается, створки клапана полностью раскрываются и кровь свободно поступает в аорту; когда левый желудочек расслабляется и наполняется артериальной кровью, притекающей из легких, створки клапана полностью смыкаются и препятствуют обратному току крови из аорты в левый желудочек. При клапанном стенозе створки клапана частично сращены между собой, и их полное открытие становится невозможным.
Естественное течение порока. Или к чему приводит аортальный стеноз?
Аортальный стеноз заставляет левый желудочек работать в режиме сверх усилий, чтобы притолкнуть кровь через сужение в аорту. Постепенно левый желудочек устает работать в таком режиме, что приводит к растяжению его стенки, увеличению полости и развитию сердечной недостаточности.
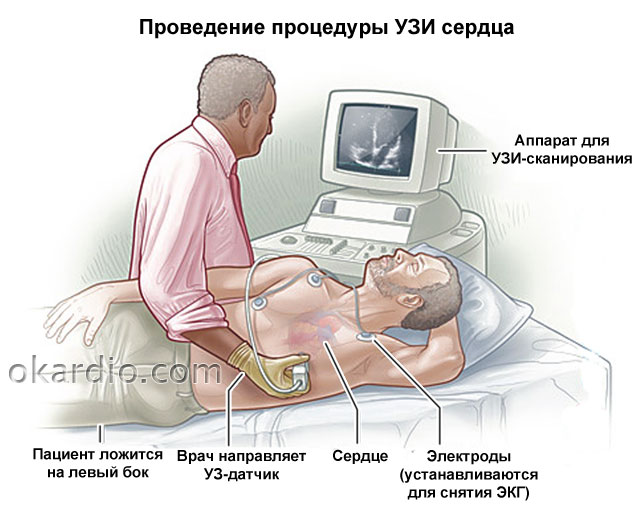
Клинические проявления аортального стеноза зависят от выраженности сужения. Так, у некоторых новорожденных с очень выраженным (критическим) стенозом, у которых еще до их рождения сердце работало против большого сопротивления, симптоматика явно выраженная. У таких детей частое дыхание и большая частота сердечных сокращений, вялость, отказ от пищи. В самых тяжелых случаях эти пациенты могут нуждаться в искусственной вентиляции легких и лечении в отделении реанимации. Им показано немедленное эндоваскулярное или оперативное лечение. Невыраженные стенозы могут не проявляться. Такие пациенты не нуждаются в лечении. Чаще всего им рекомендуют плановое наблюдение кардиолога и периодическое УЗИ сердца, которое поможет отследить прогрессирование сужения.
Лечение аортального стеноза.
|
Рис 2 – Баллоны для дилятации. Вверху в сложенном состоянии, внизу – в раздутом. |
Выбор операции зависит от места сужения. Так эндоваскулярному лечению подлежат только клапанные аортальные стенозы. Лечение клапанного аортального стеноза начинается в рентген-операционной. Такая операция называется баллонной аортальной вальвулопластикой. Через бедренную артерию в аорту под контролем рентгена вводится тонкая трубочка (катетер), через которую вводится контрастное вещество (Видео 1). Данная манипуляция позволяет определить место и степени сужения. Затем в аорту заводят катетер, на конце которого находится баллон в сложенном состоянии (Рис 2). Когда баллон достигает места сужения, его раздувают, разделяя при этом сращенные между собой створки клапана (Видео 2). Баллон сдувают, а катетер извлекают из тела пациента. С помощью другого катетера проводят измерение давления в левом желудочке и аорте и оценку эффективности процедуры. Длится данная процедура в среднем около одного часа. На теле пациента после такой операции остается только след от прокола бедренной артерии.
Реабилитация после процедуры.
Как правило, пациентов выписывают на следующий день после процедуры. Если стеноз был критическим, ребенок задерживается в клинике дольше. На месте введения катетера в сосуд еще некоторое время должна оставаться стерильная повязка. В течение шести месяцев после процедуры необходимо воздержаться от плановой вакцинации.
|
Видео 1 – Видео из операционной. Контрастное вещество вводиться через диагностический катетер в корень аорты. |
|
Видео 2 – Видео из операционной. Баллон-катетер раздувает зауженный клапан. |
Дефект межпредсердной перегородки
Различают вторичный и первичный дефект.
Вторичный локализуется в области овального окна и проявляется дефицитом
структуры в отличии от открытого овального окна. Этот вариант порока
составляет 70% от всех типов ДМПП (рис.74).
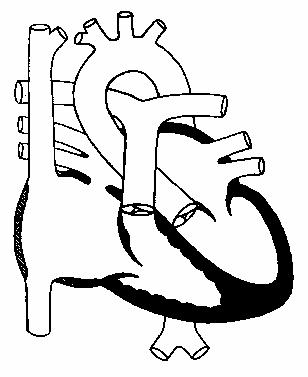
Рис.74.
Вторичный дефект
межпредсердной
перегородки (схема).
Первичный дефект локализуется в области центрального фиброзного
тела, составляет 20% от всех типов ДМПП и часто сочетается с полной
или частичной формой открытого атриовентрикулярного канала. Дефект
венозного синуса составляет 2-4% от всех ДМПП и локализуется в области
соединения межпредсердной перегородки с верхней полой веной, часто
сочетается с аномалиями легочных вен.
Одномерная ЭхоКГ:
- Дилатация правого желудочка.
- Объемная перегрузка правого желудочка.
- Увеличение экскурсии передней створки трикуспидального
клапана. - Парадоксальное (А, В типы) движение межжелудочковой
перегородки. - Преждевременное открытие клапана легочной артерии.
- Диастолическое трепетание передней створки трикуспидального
клапана
Двухмерная ЭхоКГ:
Прямая визуализация дефекта (рис.75).
- Вторичный дефект проявляется дефицитом структуры
в области овального окна.
Рис.75.
Вторичный дефект
межпредсердной
перегородки.
- Первичный ДМПП — визуализируется в
проекции 4-х камер с верхушки или из субкостального доступа в области
центрального фиброзного тела-место соединения межпредсердной перегородки
с межжелудочковой перегородкой (рис.76).
Рис.76.
Первичный дефект
межпредсердной
перегородки.
- Дефект венозного синуса — прямая
визуализация дефекта из субкостального доступа проксимально впадению
верхней полой вены в правое предсердие.
Дифференциальный диагноз:
- При допплер-ЭхоКГ необходимо дифференцировать нормальный
поток в верхней полой вене от шунтового через дефект. - Открытый атриовентрикулярный канал.
- Аномалия Эбштейна (при этом пороке дефект межпредсердной
перегородки или открытое овальное окно определяются в 85% случаев). - Триада Фалло.
Классификация
Существует множество классификаций врождённых пороков.
ВПС условно делят на 2 группы:
1. Белые (бледные, с лево-правым сбросом крови, без смешивания артериальной и венозной крови). Включают 4 группы:
С обогащением малого круга кровообращения (открытый артериальный проток, дефект межпредсердной перегородки, дефект межжелудочковой перегородки, АВ-коммуникация и т. д.).
С обеднением малого круга кровообращения (изолированный пульмональный стеноз и т. д.).
С обеднением большого круга кровообращения (изолированный аортальный стеноз, коарктация аорты и т. д.)
Без существенного нарушения системной гемодинамики (диспозиции сердца — декстро-, синистро-, мезокардии; дистопии сердца — шейная, грудная, брюшная).
2. Синие (с право-левым сбросом крови, со смешиванием артериальной и венозной крови). Включают 2 группы:
- С обогащением малого круга кровообращения (полная транспозиция магистральных сосудов, комплекс Эйзенменгера и т. д.).
- С обеднением малого круга кровообращения (тетрада Фалло, аномалия Эбштейна и т. д.).
В 2000 году была разработана Международная Номенклатура врождённых пороков для создания общей классификационной системы.
Гипоплазия
Гипоплазия может поражать сердце, как правило, приводя к недоразвитию правого или левого желудочка. Это приводит к тому, что только одна сторона сердца способна эффективно перекачивать кровь к телу и лёгким. Гипоплазия сердца встречается редко, но это наиболее серьёзная форма ВПС. Такие состояния называют синдром гипоплазии левых отделов сердца, когда поражается левая сторона сердца и синдром гипоплазии правых отделов сердца, когда поражается правая сторона сердца
При обоих состояниях, наличие открытого артериального протока (а когда гипоплазия поражает правую сторону сердца, и открытого овального окна) жизненно важно для возможности ребёнка дожить до выполнения операции на сердце, так как без этих путей кровь не сможет циркулировать в организме (или лёгких, в зависимости от стороны поражения сердца). Гипоплазия сердца, как правило, синий порок сердца.
Дефекты обструкции
Дефекты обструкции возникают, когда клапаны сердца, артерии или вены стенозированы или атрезированы. Основные пороки — стеноз лёгочного клапана, стеноз аортального клапана, а также коарктация аорты. Такие пороки как стеноз двустворчатого клапана и субаортальный стеноз возникают относительно редко. Любой стеноз или атрезия может привести к расширению сердца и гипертонии.
Дефекты перегородки
Перегородка — стенка ткани, отделяющая левое сердце от правого. При дефектах межпредсердной или межжелудочковой перегородки кровь движется из левой части сердца в правую, уменьшая эффективность работы сердца. Дефект межжелудочковой перегородки наиболее распространённый тип ВПС.
Синие пороки
Синие пороки сердца, называются так, потому что они приводят к цианозу, при этом кожа приобретает голубовато-серый цвет из-за нехватки кислорода в организме. К таким порокам относят персистирующий артериальный ствол, тотальная аномалия соединения лёгочных вен, тетрада Фалло, транспозиция магистральных сосудов, а также врождённый стеноз трёхстворчатого клапана.
Пороки
- Аортальный стеноз
- Дефект межпредсердной перегородки
- Дефект предсердно-желудочковой перегородки
- Стеноз двустворчатого клапана
- Декстрокардия
- Удвоение выходного отверстия левого желудочка
- Удвоение выходного отверстия правого желудочка
- Аномалия Эбштейна
- Синдром гипоплазии левых отделов сердца
- Синдром гипоплазии правых отделов сердца
- Стеноз митрального клапана
- Атрезия лёгочной артерии
- Врождённый стеноз клапана лёгочной артерии
-
Транспозиция магистральных сосудов
- dextro-Транспозиция магистральных сосудов
- senistro-Транспозиция магистральных сосудов
- Врождённый стеноз трёхстворчатого клапана
- Персистирующий артериальный ствол
- Дефект межжелудочковой перегородки
Некоторые состояния поражают только крупные сосуды в непосредственной близости от сердца, однако их часто классифицируют как ВПС.
- Коарктация аорты
- Атрезия аорты
- Открытый артериальный проток
- Частичная аномалия соединения лёгочных вен
- Тотальная аномалия соединения лёгочных вен
Некоторые группы пороков обычно встречаются вместе.
- тетрада Фалло
- пентада Кантрелла
- синдром Шона/ комплекс Шона / аномалия Шона
Диагностика
Диагностические мероприятия при обнаружении порока сердца и определении его типа требуют комплексного подхода. Для начала врач собирает анамнез: выясняет жалобы пациента, время и обстоятельства их проявления, интенсивность, наследственные факторы. На этом этапе ценной будет и информация от близких людей, нередко они замечают подробности, которым больной не придает значения.
Далее врач проводит физикальную диагностику:
- визуальный осмотр;
- пальпацию (ощупывание);
- перкуссию (простукивание);
- аускультацию (выслушивание).
Визуально оценивают окрас кожного покрова, строение тела пациента, особенности развития, предпочитаемую позу. При пальпации определяются характеристики пульса, сердечных сокращений, систолическое дрожание, температура кожных покровов на разных участках тела. Аускультативно определяются тоны сердца, их акценты, диастолические и систолические шумы. Перкуторно выявляется расширение границ сердца, других органов. Обязательно измеряется артериальное давление.
В обязательном порядке назначается лабораторная диагностика:
- биохимический и общий анализ крови (СОЭ, лейкоциты, гемоглобин);
- серологическое исследование (белки, белковые фракции, фибриноген и пр.);
- коагулограмма.
После физикальной и лабораторной диагностики назначают инструментальные методы обследования.
Лечение и прогнозы
Восстановить целостность межпредсердной перегородки можно исключительно инструментальным путем. Лучшим временем для проведения операции служит возрастной промежуток 1-12 лет. Отказаться от операции придется, если у больного выявлено чрезмерно повышенное внутрилегочное давление на фоне вено-артериального кровообращения, так как сосуды легких меньшего диаметра уже претерпевают склеротические изменения.
В хирургии используется несколько видов оперативных манипуляций:
- ушивание просвета;
- пластическое восстановление межпредсердного окна биологическим или синтетическим лоскутом;
- рентгенэндоваскулярное сужение при небольших ДМПП (до 2 см).
После проведенной манипуляции наблюдаются положительные прогнозы отдаленной перспективы в 90% случаев. Нормализация кровообращения по малому кругу наступает сразу, больные не высказывают жалоб на плохое самочувствие в связи с сердечным заболеванием. Если у человека присутствует небольшой дефект, он может вообще не проявить себя вплоть до пожилого возраста, не сопряжен с опасностями для жизни. Если обнаружено значительное изменение в строении перегородки, без лечения продолжительность жизни составляет 35-40 лет, летальный исход может наступить от дыхательных проблем или чрезмерной гипертензии.
Статья размещена с целью информирования без претензии на научную точку зрения. Если у Вас появились подозрения о наличии заболевания, обратитесь к кардиологу или кардиохирургу, пройдите обследование.
Симптомы и признаки дефекта межпредсердной перегородки
Признаки, характерные для пороков сердца, такие как одышка, сердцебиение, быстрая утомляемость при физической нагрузке обычно впервые появляются к третьему году жизни ребенка. Ребенок отстает в физическом развитии, по возможности, уклоняется от занятий физкультурой, часто болеет воспалением легких. Точная картина заболевания и сроки появления жалоб напрямую зависят от размеров дефекта. В легких случаях до появления изменений в состоянии здоровья может пройти несколько десятилетий. Ухудшение состояния пациентов связано с возникающими нарушениями сердечного ритма или развивающейся сердечной недостаточностью.
Причины
Большинство случаев дефектов межпредсердной перегородки возникают сами по себе без видимой причины (спорадически). Точная природа развивающего порока или дефектов, которые могут возникнуть во время эмбрионального развития (эмбриогенеза), остается неясной.
Некоторые случаи ДМПП проявляются в семьях. В таких редких случаях два типа, ostium primum и ostium secundum, по-видимому, наследуются как аутосомно-доминантные генетические признаки. Чтобы еще больше усложнить ситуацию, генетический анализ предполагает, что есть как минимум два различных генетических нарушения, связанных с ДМПП, которые связаны с мутациями в гене, называемом Nkx2-5.
Хромосомы, которые присутствуют в ядре клеток человека, несут генетическую информацию для каждого человека. Клетки человеческого тела обычно имеют 46 хромосом. Пары человеческих хромосом пронумерованы от 1 до 22, а половые хромосомы обозначены X и Y. У мужчин есть одна Х и одна Y-хромосома, а у женщин две Х-хромосомы. Каждая хромосома имеет короткое плечо, обозначенное «p», и длинное плечо, обозначенное «q». Хромосомы далее подразделяются на множество пронумерованных полос. Например, «хромосома 6p21.3» относится к полосе 21.3 на коротком плече хромосомы 6. Аналогично, «хромосома 8p23.1-p22» относится к области между полосами 22 и 23.1 на коротком плече хромосомы 8. Нумерованные полосы указывают местоположение тысяч генов, которые присутствуют на каждой хромосоме.
Генетические заболевания определяются сочетанием генов для определенного признака, которые находятся на хромосомах, полученных от отца и матери.
Доминантные генетические нарушения возникают, когда для появления заболевания необходима только одна копия ненормального гена. Аномальный ген может быть унаследован от любого из родителей или может быть результатом новой мутации (изменения гена) у пострадавшего человека. Риск передачи ненормального гена от пострадавшего родителя к потомству составляет 50% для каждой беременности, независимо от пола ребенка.
Рецессивные генетические нарушения возникают, когда человек наследует один и тот же аномальный ген по одному признаку от каждого родителя. Если человек получает один нормальный ген и один ген заболевания, человек будет носителем заболевания, но обычно бессимптомным. Риск того, что двое родителей-носителей оба передадут дефектный ген и, следовательно, заведут больного ребенка, составляет 25 процентов при каждой беременности. Риск родить ребенка, который будет являться носителем, как родители, составляет 50 процентов с каждой беременностью. Вероятность для ребенка получить нормальные гены от обоих родителей и быть генетически нормальным для этой конкретной болезни составляет 25 процентов. Риск одинаков для мужчин и женщин.
Все люди несут 4-5 ненормальных генов. Родители, которые являются близкими родственниками (брат и сестра), имеют более высокий шанс, чем несвязанные родители, иметь один и тот же ненормальный ген, что повышает риск рождения детей с рецессивным генетическим расстройством.
Дефект межпредсердной перегородки может также возникать в связи с множеством других врожденных пороков сердца, или у новорожденных, которые являются относительно маленькими или преждевременными. Первичная форма болезни часто встречаются у людей с синдромом Дауна или синдромом Эллиса ван-Кревельда.
Общая информация о болезни
Дефект межпредсердной перегородки (ДМПП) представляет собой редкий врожденный порок сердца, характеризующийся отверстием в стенке (перегородке), разделяющей две верхние камеры (предсердия) сердца.
Обычно сердце имеет четыре камеры: две верхние камеры, известные как предсердия, отделенные друг от друга волокнистой перегородкой, известной как перегородка предсердия, и две нижние камеры, известные как желудочки, отделенные друг от друга перегородкой желудочка.
Клапаны соединяют предсердия (левый и правый) с соответствующими желудочками. При рождении между двумя предсердиями присутствует небольшое отверстие (овальное отверстие). Вскоре после рождения межпредсердная перегородка постепенно растет и закрывает это отверстие. У детей с ДМПП межпредсердная перегородка может не закрыться должным образом или повредится во время развития плода. При этом нарушении (называемом открытым овальным отверстием) между предсердиями долгое время сохраняется отверстие, хотя она должна быть закрыто, что приводит к увеличению рабочей нагрузки на правую сторону сердца и чрезмерному притоку крови к легким.
Первоначально симптомы, связанные с дефектом межпредсердной перегородки, могут отсутствовать или быть настолько слабовыраженными, что могут оставаться незамеченными. Часто расстройство не распознается до школьного возраста или даже до зрелого возраста. У взрослых с необнаруженным дефектом межпредсердной перегородки могут развиться различные проблемы с дыханием и/или сердечная недостаточность.
Выявлены несколько форм ДМПП. Они классифицируются в зависимости от их расположения в перегородке. Первичная форма (тип ostium primum) относится к дефекту, находящемуся в нижней части перегородки. Вторичная форма (тип ostium secundum) относится к дефекту, расположенному в середине перегородки, а форма sinus venosus (дефект венозного синуса) относится к дефектам в верхней части перегородки.
Что такое вторичный дефект межпредсердной перегородки?
Вторичные дефекты встречаются чаще, чем первичные. Конкретное местонахождение отверстия в этом случае бывает различным, но всегда сохраняется край межпредсердной перегородки на границе с предсердно-желудочковыим отверстием.
Читайте больше о кардиологическом отделении больницы Шиба — http://www.hospital-direct.org.il/cardiology-department.aspx.
В зависимости от точной зоны дислокации дефекта, он классифицируется как центральный, нижний, верхний, задний, передний, а также встречаются множественные дефекты. Вторичный дефект межпредсердной перегородки может быть как самостоятельным пороком сердца, так и встречаться в сочетании с другими его пороками, например, при нарушении развития венозного синуса или при наличии аномального дренажа легочных вен справа и верхней полой вены.
Размер дефекта, имеющий определяющее значение на состояние больного, может варьировать у разных пациентов в очень широких пределах — от 1 см до, практически, полного отсутствия межпредсердной перегородки.
Примерно у 15% детей, родившихся со вторичным дефектом межпредсердной перегородки, это отверстие может закрываться на первом году жизни. Но если этого не произошло, то в более поздние сроки самостоятельное закрытие, практически, не наблюдается. Поскольку сердечнососудистая система человека обладает довольно большими компенсаторными возможностями, то патологические изменения в ней при этом пороке развиваются довольно медленно. Симптомы болезни у детей первого года жизни наблюдаются лишь в случае наличия больших дефектов. Количество таких больных составляет не более 1%. Поскольку патологический механизм нарушения кровообращения выражен перемещением части крови из левого предсердия в правое, то наблюдается перегрузка правых отделов сердца. Сначала развивается гипертрофия правого желудочка, в дальнейшем к ней присоединяется повышение давления в легочной артерии. Следующим этапом развития болезни служат изменения, появляющиеся в легких. Как правило, это происходит в возрасте около16 лет.
Тетрада Фалло и беременность
ОПИСАНИЕ
Тетрада Фалло – это врожденный порок сердца представленный дефектом межжелудочковой перегородки, аортой «наездником» (декстрапозиция), стенозом легочной артерии и гипертрофией миокарда правого желудочка. Тетрада Фалло относится к наиболее распространенным порокам сердца синего типа, встречается в 10 -12% от всех врожденных пороков сердца. Частота порока сердца у потомства составляет примерно 5%.
ПРИЧИНЫ
Основной причиной развития заболевания служит нарушение генома. Генетики относят его к понятию синдром diGeorge (Диджорджи или Ди Георге).
Причина развития синдрома кроется в субмикроскопической делеции (отщеплении) района q11.2 хромосомы 22. Сокращенно этот вариант патологии генетики пишут так: «del 22 q11.2». Но интересно то, что подобное нарушение генотипа проявляется не только патологией развития сердца в виде тетрады Фалло. Делеция этого участка имеет также обозначения DGCR (DiGeorge syndrome critical region) и VCF(velocardiofacial syndrome).
На уровне организма при этом встречаются проявления со стороны различных органов и систем. Обычно синдром сочетает гипокальциемию (за счет недоразвития паращитовидных желез), Т-клеточный иммунодефицит (из-за недоразвития тимуса), пороки выходных отверстий сердца (к которым относят и тетраду Фалло), а также лицевые мальформации. В связи с тем, что тетрада Фалло является лишь одним из вариантов пороков, связанных с «del 22», вполне резонно предположить, что данный генетический дефект встречается не реже, чем тетрада Фалло. И действительно, синдром diGeorge встречается примерно у одного из 3000 новорождённых.
СИМПТОМЫ
Цианоз – основной симптом тетрады Фалло. Степень цианоза и время его появления зависит от выраженности стеноза лёгочной артерии. В основном, цианоз развивается к 3 месяцам, к 1 году.
Цианоз нарастает с ростом активности ребенка. Постоянным признаком порока является одышка, которая резко возрастает при малейшей физической нагрузке. В тяжелых случаях при тетраде Фалло, развиваются приступы удушья. Возникают они, как правило, в возрасте от 6 до 24 месяцев. Ребенок становится беспокойным, выражение лица испуганное, зрачки расширены, одышка и цианоз нарастают, конечности холодные; затем следует потеря сознания, судороги и возможно развитие комы и летальный исход.
Приступы длятся обычно несколько минут. Постепенно развивается задержка физического развития, пациенты часто имеют пальцы рук и ног в виде «барабанных палочек». Практически с рождения выслушивается грубый систолический шум вдоль левого края грудины.
Осложнения при беременности
До беременности рекомендуется хирургическое лечение порока сердца. Пациенты с полной коррекцией порока и отсутствием сердечной недостаточности беременность переносят хорошо, хотя в некоторых случаях могут иметь место нетяжелые нарушения ритма сердца. При некоррегированном пороке имеется высокий риск материнской смертности, невынашивания беременности и внутриутробной задержки роста плода. При некоррегированном пороке беременность противопоказана.
Факторы риска неблагоприятного исхода беременности:
- высокий гематокрит, превышающий 65%;
- застойная сердечная недостаточность;
- увеличение размеров сердца;
- насыщение крови кислородом менее 80%.
ЛЕЧЕНИЕ
Выделяют радикальные и паллиативные способы коррекции порока.Радикальная ликвидация порока проводится в условиях кардиоплегии и искусственного кровообращения. Операция заключается в закрытии межжелудочкового дефекта и ликвидации стеноза.
Паллиативные операции содержатся в наложении окольных межартериальных анастомозов. Задача операции — предоставить детям шанс пережить тяжелый период, для того, чтобы потом провести радикальную операцию.
1.Общие сведения
Аневризма (изредка употребляется менее корректный термин «аневризм») – локальное выбухание, выпячивание какой-либо граничной поверхности в виде растянутого мешка, либо веретенообразное расширение стенок кровеносного сосуда (мешотчатые аневризмы также встречаются на сосудах, однако значительно реже). Таким образом, аневризма межпредсердной перегородки (МПП) – это искривление мембраны, отделяющей правое предсердие от левого. В норме эта перегородка герметична, сравнительно пряма и не выгибается, – по крайней мере, существенно, – ни в одну из сторон.
Аневризма МПП многими авторами квалифицируется как патология лишь в том случае, если выпячивание в какую-либо сторону (в правое предсердие, в левое или S-образно) превышает 10 мм и сочетается с пролапсом (провисанием) митрального клапана. В пересчете к общей популяции такая аномалия встречается редко, чаще она сопутствует врожденным заболеваниям соединительной ткани. Аневризму МПП называют «малой сердечной аномалией» или «малым пороком сердца», поскольку в большинстве бессимптомных случаев она не представляет реальной опасности и не нуждается в лечении, да и обнаруживается, как правило, случайно. Однако из этого отнюдь не следует, что о данной аномалии можно забыть, не придавая ей никакого значения: ситуация может резко усугубиться с началом увеличения аневризмы, истончения МПП и ее спонтанного прорыва, после чего динамика становится уже непредсказуемой.
Недостаточность митрального клапана (НМК)
Неспособность левого предсердно-желудочкового клапана препятствовать обратному току крови из левого желудочка в левое предсердие во время систолы (сокращения) желудочков сердца. Регистрируется у 50% больных с различными пороками сердца, но в чистом виде встречается редко. У детей наблюдается гораздо чаще, чем у взрослых. Часто совмещается с митральным стенозом или пороками клапанов аорты. В течении НМК условно выделяют 3 периода: компенсации, легочной венозной гипертензии и правожелудочковой недостаточности.
Поскольку компенсация приходится на самый мощный отдел сердца – левый желудочек, то этот период довольно длительный. Бессимптомное течение можно наблюдать на протяжении нескольких лет. При появлении клинических симптомов наиболее типичны жалобы на одышку (98%), быструю утомляемость (87%). На более поздних стадиях отмечаются:
- акроцианоз – цианоз губ, кончика носа, пальцев;
- ортопноэ – одышка в лежачем положении, пациент вынужденно предпочитает сидячее либо полулежащее положение;
- отеки на ногах;
- набухание шейных вен;
- редко – увеличение живота в объеме (асцит).