Синдром патау (трисомия по 13 хромосоме) и синдром эдвардса (трисомия по 18 хромосоме). анеуплоидия, трисомия, транслокация, мозаицизм
Содержание:
Причины
Трисомия 18 развивается как следствие наличия дополнительной 18-й хромосомы в кариотипе зиготы. Возникновение этой патологии случайно, и предотвратить его нельзя. Но определяются некоторые факторы риска развития этой патологии:
- Возраст матери – чем старше женщина, тем выше ожидаемый риск трисомии. Вероятность развития болезни Эдвардса увеличивается после 35 лет – с каждым годом частота проявления таких мутаций у ребенка выше.
- Генетическая расположенность – вероятность повышается, если в семье уже были случаи такого заболевания.
- Прием некоторых препаратов, влияющих на деление клеток эмбриона.
- Перенесенные инфекционные заболевания.
- Курение и потребление алкоголя.
Показания для тестирования на ломкую Х-хромосому
- Индивиды с задержкой умственного и общего развития, аутизмом
- Индивиды с чертами фрагильной Х-хромосомы
- Индивиды с наличием синдрома фрагильной Х-хромосомы в семейном анамнезе
- Индивиды с наличием в семейном анамнезе недиагностированной задержки умственного развития
- Плоды от матерей-носителей
Геномный импринтинг — процесс, при котором активация гена происходит преимущественно в материнской или преимущественно в родительской хромосоме, но не в обеих хромосомах. Нормальное развитие имеет место лишь в том случае, если присутствуют обе копии (материнская и отцовская) импринтинг-ген. Импринтинг-ген неактивен, значит, активный ген теряет (путем делеции) или получает мутацию, в таком случае плод будет пораженным. Лишь несколько генов могут испытывать импринтинга. Примерами геномного импринтинга может быть синдром Ангельмана и полный пузырный занос (вариант гестационной трофобластической болезни).
Синдром Ангельмана характеризуется тяжелой задержкой умственного развития, атаксической походкой, типичным лицом, пароксизмами смеха и судорогами. Ген синдрома Ангельмана является активным только в материнской унаследованной хромосоме, следовательно, если происходит делеция материнской хромосомы 15 или материнская копия гена имеет мутацию, белковый продукт не образуется и плод будет пораженным.
Синдром Ангельмана также может развиться, если обе копии хромосомы 15 является унаследованными от отца (отсутствие материнской копии хромосомы 15). Это состояние получило название унипарентальной дисомии. Унипарентальная дисомия возникает чаще вследствие потери хромосомы у эмбриона с трисомией или добавления хромосомы у плода с моносомией по этой хромосомой. Каждая из хромосом может быть генетически различной (гетеродисомия) или идентичной (изодисомия), в зависимости от времени возникновения этого феномена — в течение первого или второго мейотического деления, соответственно.
Полный пузырный занос обычно является диплоидным (46, ХХ или Х ¥), но может иметь полностью отцовское происхождение, без материнского хромосомного материала. При таких условиях плод не может развиваться. Полный пузырный занос может сопровождать нормальную многоплодную беременность, но в этом случае возрастает риск материнских осложнений (гипертиреоидизм, преэклампсия, преждевременные роды). В отличие от полного пузырного заноса, частичный пузырный занос обычно является триплоидным (69, ХХХ, 69, ХVV), с дополнительным набором отцовских хромосом.
Триплоидия с дополнительным набором материнских хромосом имеет место при ЗВУР плода, врожденных пороках развития и маленькой плаценте.
Синдром Эдвардса — симптомы
Обычно синдром Эдвардса выявляют во время беременности с помощью специальных пренатальных тестов. Это дефект, который обычно вызывает самопроизвольный выкидыш на ранних сроках беременности. Однако, если беременность сохраняется, ребенок рождается с набором дефектов и симптомов, которые возникают в результате наличия дополнительной хромосомы 18.
Эти дефекты могут иметь различную степень тяжести, и это зависит, прежде всего, от типа генетического нарушения (есть ли только дополнительный фрагмент или целая хромосома). Наиболее частые симптомы включают:
- Низкий вес при рождении.
- Микроцефалия.
- Расщелина губы или неба.
- Маленькая нижняя челюсть.
- Широкое расстояние между глазами (гипертелоризм), опущение верхнего века.
- Деформация стопы.
- Большие пальцы рук и / или ногти не полностью развиты.
- Нет лучевой кости.
Расщелина губы
Что такое анеуплоидия, трисомия, транслокация, мозаицизм
В каждой клетке человеческого организма находится 46 хромосом, в которых выделяют две группы: 22 пары аутосом (пронумерованных с 1 по 22, в зависимости от размера) и пара половых хромосом (XX у женщин, XY у мужчин). Каждая хромосома в паре является гомологичной другой хромосоме в паре.
В норме человек имеет диплоидный набор хромосом, то есть в каждой клетке содержится двойной комплект каждой из 23 хромосом.
Но есть ситуации, в которых клетки содержат ненормальный, не кратный 46, набор хромосом, что называется анеуплоидией. Анеуплоидия может выражаться, например, в наличии добавочной хромосомы (n + 1, 2n + 1 и т. п.) или в нехватке какой-либо хромосомы (n — 1, 2n — 1 и т. п.).
Формы анеуплоидии:
- моносомия (наличие одной из пары хромосом, например, синдром Шерешевского-Тернера, выражающийся в наличие одной половой Х-хромосомы)
- трисомия (наличие трех вместо 2 хромосом пары).
- тетрасомия (4 гомологичные хромосомы вместо пары в диплоидном наборе)
- пентасомия (5 вместо 2-х) встречаются чрезвычайно редко.
Дальше речь пойдет о самых частых хромосомных аномалиях — трисомиях. В некоторых случаях дополнительная хромосома представлена целой отдельной хромосомой (полная трисомия), а в некоторых этот генетический материал переносится на другую хромосому, что называют транслокацией.
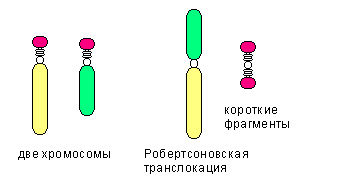
Среди транслокаций также выделяют:
- реципроктную транслокацию, когда неготомологичные хромосомы обмениваются участками
- робертсоновскую транслокацию (см.рис), при которой две неготомологичные хромосомы объединяются в одну.
- Сбалансированная транслокация не сопровождается утратой генетического материала.
Мозаицизмом называют ситуацию, когда среди всех клеток организма есть нормальные, а есть клетки с патологией (например, с трисомией). В этом случае степень отклонений зависит от количества клеток, которые имеет ненормальный генетический материал.
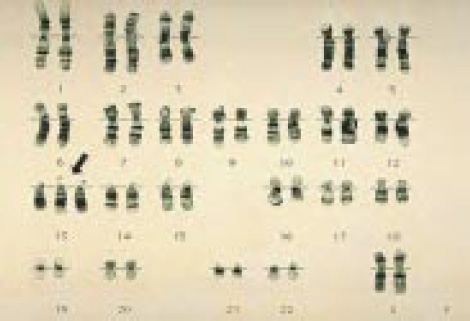
Хромосомы в случае синдрома Патау — Трисомия 13

Хромосомы в случае синдрома Эдвардса — Трисомия 18
Общие сведения
Синдром Эдвардса (заболевание также называют синдром трисомии 18) – это хромосомная болезнь, для которой характерны множественные пороки развития и трисомия 18 хромосомы. Впервые эту болезнь описал Джон Эдвардс в 1960 году. Диагностируется такое заболевание редко – как свидетельствует статистика, в мире оно встречается с частотой 1:5000. Отмечено, что дети с такой патологией чаще рождаются у матерей зрелого возраста. Так, вероятность появления на свет малыша с такой патологией для женщины старше 45 лет составляет 0,7 %. Риск увеличивается, если будущая мать болеет сахарным диабетом. При этом девочки с такой патологией появляются на свет в три раза чаще, чем мальчики.
Болезнь Эдвардса – очень тяжелая патология, при которой выживаемость после года составляет 5–10%. Как диагностируется это заболевание и с чем связано его развитие, речь пойдет в этой статье.
Влияние хромосомных аберраций на развитие
Большой вклад в дело выяснения масштаба проблемы внесли исследования Альфреда Гроппа из Любека и Чарльза Форда из Оксфорда, проводившиеся на так называемых «табачных мышах» (Mus poschiavinus). Скрещивание подобных мышей с нормальными мышами дает большой спектр триплоидий и моносомий, позволяющих оценить влияние обоих типов аберраций на развитие.
Данные профессора Гроппа (1973 г.) приведены в таблице.
| Распределение эуплоидных и анэуплоидных зародышей у гибридных мышей | |||||
|---|---|---|---|---|---|
| Стадия развития | День | Кариотип | Всего | ||
| Моносомии | Эуплоидии | Трисомии | |||
| До имплантации | 4 | 55 | 74 | 45 | 174 |
| После имплантации | 7 | 3 | 81 | 44 | 128 |
| 9—15 | 3 | 239 | 94 | 336 | |
| 19 | 56 | 2 | 58 | ||
| Живые мыши | 58 | 58 |
Эти исследования позволили подтвердить гипотезу о равной вероятности возникновения моносомий и трисомий при зачатии: аутосомные моносомии возникают с такой же частотой, как и трисомии, но зиготы с аутосомными моносомиями погибают еще до имплантации и не обнаруживаются в материале выкидышей.
При трисомиях гибель зародышей происходит на более поздних сдадиях, но ни один зародыш при аутосомных трисомиях у мышей не доживает до родов.
Исследования группы Гроппа позволили показать, что в зависимости от типа трисомии, зародыши погибают на разных сроках: с трисомиями 8, 11, 15, 17 — до 12 дня после зачатия, с трисомиями 19 — ближе к сроку родов.
Как выглядят новорожденные с синдромом Эдвардса?
Новорожденные с синдромом Эдвардса имеют следующие характерные аномалии развития:
- изменение формы черепа;
- изменение формы ушных раковин;
- аномалии развития неба;
- стопа-качалка;
- аномальная длина пальцев;
- изменение формы нижней челюсти;
- сращение пальцев;
- аномалии развития половых органов;
- флексорное положение кистей;
- дерматоглифические признаки.
Аномалии развития неба
его можно прощупать посередине твердого неба языкомСуществует несколько вариантов данного дефекта:
- незаращение мягкого неба (задняя, глубокая часть неба, которая нависает над глоткой);
- частичное незаращение твердого неба (щель не тянется на протяжении всей верхней челюсти);
- полное незаращение твердого и мягкого неба;
- полное незаращение неба и губы.
даже при закрытом ртепочти 20% новорожденныхдо 65% новорожденных
Изменение формы нижней челюсти
микрогенияУ новорожденных с микрогнатией обычно быстро появляются следующие проблемы:
- невозможность долго держать рот закрытым (подтекание слюны);
- затруднения при кормлении;
- позднее развитие зубов и неправильное их расположение.
Дерматоглифические признаки
аномальные узоры и складки на коже ладонейОсновными дерматоглифическими признаками синдрома Эдвардса являются:
- дуги на подушечках пальцев располагаются с большей частотой, нежели у здоровых людей;
- кожная складка между последней (ногтевой) и предпоследней (срединной) фалангами пальцев отсутствует;
- у 30% новорожденных на ладони имеется так называемая поперечная борозда (обезьянья линия, линия Симиан).
Аномалии половых хромосом
Наиболее частыми аномалиями половых хромосом является синдром Тернера (моносомия Х, или 45, Х0, мозаицизм) и синдром Кляйнфельтера (47, ХХУ). Это может быть обусловлено тем обстоятельством, что кариотипы 47, ХХХ и 47, ХVV не имеют выраженных фенотипических различий и поэтому идентифицируются реже.
Синдром Тернера (Шерешевского — Тернера) может быть вызван потерей родительской хромосомы, мозаицизмом (45, Х / 46, ХХ или 45, Х / 46, ХУ) или структурными аномалиями Х-хромосомы (делеции, изохромосомы).
Индивиды с синдромом Тернера имеют женский фенотип, дисгенезию гонад, низкий рост, первичные, отсутствие вторичных половых признаков, крыловидные складки шеи, низко расположенные уши, низкую заднюю границу роста волос, дискообразную грудную клетку с широкой расстоянием между сосками, аномалии почек, лимфедему конечностей при рождении и кардиоваскулярные аномалии, чаще коарктацию аорты. Но при ультразвуковом исследовании может проявляться только одна аномалия — кистозная гигрома. Скрининг-теста синдромом Тернера еще не существует, и поэтому частота рецидивов не определена.
В случае синдрома Кляйнфельтера развитие яичек сначала является нормальным. Но присутствие не менее 1 лишней Х-хромосомы приводит к гибели зародышевых клеток на этапе их поступления в мейоз, что приводит к уменьшению яичек и гиалинизации семенных протоков. Итак, классические симптомы синдрома Кляйнфельтера включают бесплодие, гинекомастию, задержку умственного развития, повышения уровня гонадотропинов вследствие уменьшения уровня циркулирующих андрогенов. Скрининг-теста по выявлению синдрома Кляйнфельтера также не существует, следовательно, пренатальный диагноз этих хромосомных аномалий возможно только при использовании биопсии хориона или амниоцентеза.
Генетическая консультация
Когда проводится пренатальная или неонатальная диагностика синдрома Эдвардса, консультирование семьи должно быть реалистичным.
Родителям может быть трудно согласиться с отсутствием уверенности ситуации, но они должны быть подготовлены как к вероятности смерти, так и к возможности жить. Поскольку родители должны принимать практические решения в отношении реанимации, хирургии, жизнеобеспечения, следует объяснить все варианты спасения новорожденного.
Риск рецидива для семьи с ребенком с полной трисомией 18 обычно указывается как 1%. Сообщалось о повторении различных трисомий в одном и том же семействе.
Риск рецидива составляет менее 1%, но выше, чем возрастной. Риск повторения в семьях с частичной выше по сравнению с полной. Он зависит от наличия геномной перегруппировки (транслокации или инверсии) у одного из родителей.
Лечение синдрома
К сожалению, излечение данного синдрома невозможно.
Терапия, которая может проводиться, обычно направлена на снятия остроты проявляющихся симптомов болезни и предотвращение возникновения сопутствующих заболеваний, составляющих действительно огромный список:
- пневмония, хронический бронхит, астма;
- хронический отит;
- онкологические заболевания разных органов (например, рак горла);
- синусит;
- повышенное или пониженное давление.
Можно отметить, что большинство сопутствующих заболеваний приобретает хронический характер. Чтобы этого не происходило, врачи стараются применять разного рода терапию, если это возможно.
Медикаментозное лечение
Медикаментозное лечение наиболее актуально при синдроме Эдвардса у новорожденных. В терапию включают антибактериальные, противовоспалительные и, чаще всего, гормональные препараты. Они предотвращают развитие некоторых заболеваний, облегчая жизнь ребенку.
Гормональная терапия улучшает функционирование щитовидной железы в организме, что тоже немаловажно для пациента с таким диагнозом
Хирургия
Хирургическое вмешательство при столь серьезной болезни считается нерациональным, так как это может привести к осложнениям или критическому состоянию. Поэтому врачи проводят хирургическое лечение при трисомии 18 только в случае осложнения каких-либо других заболеваний у больного ребенка.
Домашние средства
Учитывая тот факт, что даже медикаментозное лечение данной болезни неэффективно, говорить о лечении дома практически не имеет смысла. Родители могут изготавливать отвары из различных травяных сборов в случае повышенной эмоциональности или агрессивности ребенка. Это поможет ему успокоиться.
Кроме того, часто используются отвары, способные укрепить иммунитет больного ребенка, который ввиду своей болезни, становится очень уязвим к вирусным заболеваниям.
Триплоидии
Крайне редко наблюдаемые при мертворождениях, триплоидии составляют пятую по частоте хромосомную аномалию в материале выкидыше. В зависимости от соотношения половых хромосом может быть 3 варианта триплоидий: 69XYY (самая редкая), 69, XXX и 69, XXY (самая частая). Анализ полового хроматина показывает, что при конфигурации 69, XXX чаще всего обнаруживается только одна глыбка хроматина, а при конфигурации 69, XXY чаще всего половой хроматин не обнаруживается.
Приведенный ниже рисунок иллюстрирует различные механизмы, приводящие к развитию триплоидии (диандрию, дигинию, диспермию). С помощью специальных методов (хромосомные маркеры, антигены тканевой совместимости) удалось установить относительную роль каждого из этих механизмов в развитии триплоидии у зародыша. Оказалось, что на 50 случаев наблюдений триплоидия была следствием дигинии в 11 случаях (22%), диандрии либо диспермии — в 20 случаях (40%), диспермии — в 18 случаях (36%).
| Механизмы образования триплоидной зиготы |
|---|
Прогноз
Абсолютное большинство детей с синдромом Эдвардса гибнет еще в утробе матери из-за того, что организм женщины отторгает плод. В случае, если ребенок родился, прогноз очень неутешителен. Большая часть детей с таким заболеванием живет до нескольких месяцев.
В том случае, если ребенок рождается не с самой серьезной формой заболевания, вероятность прожить до 10 лет резко увеличивается.
Если ребенок прожил уже несколько лет с синдромом Эдвардса, за ним можно заметить полную неактивность и слабо разбитые умственные способности. К сожалению, сделать с этим ничего нельзя, независимо от того, как за ним ухаживают родственники.
Многие родители стараются чему-то обучить своего больного ребенка – это абсолютно бесполезно. Даже тот небольшой процент больных, который достигает довольно зрелого возраста при этой болезни, умеет совсем немного.
В большинстве случаев такие люди могут самостоятельно поднимать голову, узнавать только несколько человек из ближайшего круга (те, кто чаще всего находится рядом), самостоятельно есть, иногда – передвигаться.
Если ребенок или уже взрослый человек начинает ходить, он сильно выгибает ступни внутрь, «косолапит», как говорят.
Стоит отметить, что не для всех детей подходит практика поддержания мышечного тонуса. Некоторым больным это помогает улучшить общее состояние; другим это только навредит ввиду того, что у них могут возникнуть проблемы с опорно-двигательным аппаратом и боли в позвоночнике.
По этой причине необходимо консультировать с лечащим врачом ребенка, чтобы не навредить ему.
Трисомии
В материале выкидышей трисомии представляют более половины всех количественных хромосомных аберраций
Обращает на себя внимание то, что в случаях моносомий недостающей хромосомой обычно оказывается X-хромосома, а в случаях избыточных хромосом, дополнительная хромосома чаще всего оказывается аутосомой
Точная идентификация дополнительной хромосомы стала возможна благодаря методу G-бэндинга. Исследования показали, что все аутосомы могут принимать участие в нон-дисджанкшн (см. таблицу)
Обращает на себя внимание, что три хромосомы, чаще всего встречающиеся при трисомиях новорожденных (15-я, 18-я и 21-я) чаще всего обнаруживаются и при летальных трисомиях у зародышей. Вариации относительных частот различных трисомий у зародышей отражают во многом сроки, на которых происходит гибель зародышей, поскольку, чем более летальной является комбинация хромосом, тем на более ранних сроках происходит остановка развития, тем реже будет обнаруживаться такая аберрация в материалах выкидышей (чем меньше срок остановки развития, тем труднее обнаружить такой зародыш)
| Лишняя хромосома при летальных трисомиях у зародыша (данные 7 исследований: Буэ (Франция), Карр (Канада), Кризи (Великобритания), Дилл (Канада), Кадзи (Швейцария), Такахара (Япония), Теркелсен (Дания)) | ||
|---|---|---|
| Дополнительная аутосома | Количество наблюдений | |
| A | 1 | |
| 2 | 15 | |
| 3 | 5 | |
| B | 4 | 7 |
| 5 | ||
| C | 6 | 1 |
| 7 | 19 | |
| 8 | 17 | |
| 9 | 15 | |
| 10 | 11 | |
| 11 | 1 | |
| 12 | 3 | |
| D | 13 | 15 |
| 14 | 36 | |
| 15 | 35 | |
| E | 16 | 128 |
| 17 | 1 | |
| 18 | 24 | |
| F | 19 | 1 |
| 20 | 5 | |
| G | 21 | 38 |
| 22 | 47 |
Роль 3D УЗИ при перинатальном скрининге: 3Д или 2Д?
На сегодняшний день уже ни у кого не вызывает сомнений, что методики ультразвуковой диагностики — важнейший аспект контроля внутриутробного развития, возраста и положения плода. При этом техническая база медицины не стоит на месте и в дополнение к классическому, двухмерному УЗИ пришла такая, во всех отношениях полезная диагностическая практика, как 3D УЗИ или трехмерная эхография. Преимущества этого способа исследования заключаются в возможности получения объемного изображения, которое во всех красках демонстрирует специалисту и будущим родителям, все аспекты внешних проявлений и органов малыша.
Отличия 3D УЗИ от классического ультразвукового исследования
Все ультразвуковые способы исследования имеют общий принцип, который основан на использовании ультразвукового излучения, частота волн которого не превышает 20 кГц. Подача такой волновой нагрузки в импульсном режиме позволяет оценить функциональную нормальность и морфологическое строение тканей, органов и систем плода. При этом традиционный двухмерный способ выдает на монитор приборной панели плоское изображение, которое понятно врачам, но не обладает информативностью для непрофессионалов, а именно для родителей ребенка, которые с нетерпением ждут первого знакомства с крохой
При этом следует отметить, что такой способ диагностического контроля важен для медицинских специалистов, ведущих беременность, так как он дает возможность полноценно оценить строение внутренних органов плода, что невероятно важно при организации комплексного контроля
Трехмерная эхография выдает полноценное объемное изображение, которое не требует расшифровки и отчетливо отражает внешние особенности и положение малыша в утробе матери.
Преимущества 3Д УЗИ при перинатальном скрининге
3D УЗИ предоставляет медикам ряд очевидных преимуществ:
- Более четкое изображение дает возможность установить ряд пороков, которые невозможно выявить при проведении классического ультразвукового скрининга: аномалии кистей, расщелины лица, пороки развития скелета, нарушения формирования передней брюшной стенки, аномалии последа, особенности строение наружных половых органов, незаращивание спинного мозга и т.д. Выявление всех перечисленных отклонений требует изменения стратегии ведения подобной беременности.
- 3D УЗИ позволяет определить пол малыша более точно на ранних сроках беременности, что может быть необходимо не только для удовлетворения любопытства будущих родителей, но и с точки зрения исключения вероятности наследственных патологий, связанных с половым признаком.
- Психологическая готовность матери и отца к рождению долгожданного чада, безусловно, возрастает после первичного знакомства с малышом, пусть даже посредством монитора и фотографии, которая по желанию родителей может быть предоставлена после прохождения данной манипуляции.
Особенности трехмерной эхографии
Согласно результатам многочисленных медицинских исследований, 3D УЗИ абсолютно безопасный способ диагностики, который применяется по медицинским показаниям. К особенностям проведения УЗИ 3Д при скринингах во время беременности можно отнести следующие факторы:
- Наиболее информативна трехмерная эхография на сроке 22-33 недели беременности, так как в этот период внешние признаки плода уже достаточно сформированы, а его размеры не препятствуют визуальному обзору.
- Продолжительность трехмерного УЗИ составляет около 40 минут, что гораздо дольше, чем временной показатель, необходимый для проведения классического двухмерного скрининга.
- Мочевой пузырь перед проведением 3D УЗИ не обязательно должен быть наполнен.
- Диагностические возможности методики существенно падают при наличии таких особенностей пациентки или течения беременности, как выраженное ожирение будущей матери, маловодие, наличие рубцов на брюшной стенке женщины, неудобное положение плода.
Трехмерная эхография – диагностическая практика, которая заслужила доверие врачей и пациентов всего мира, подтвердив свою исключительную эффективность и безопасность, как для женщины, так и для малыша. При этом, на сегодняшний день, именно 3D УЗИ остается «золотым стандартом» внутриутробного изучения строения лицевых структур, конечностей, половых признаков и объемных образований у плода, а также резервным способом выявления таких хромосомных аномалий как синдром Дауна, Патау и т.д.
Скрининг на генетические заболевания
Сегодня известно более 11 000 моногенных заболеваний, которые кодируются одним геном (генетически обусловленные) и передаются от родителей их потомкам. Механизм передачи многих генетических болезней объясняется принципами Менделя.
Аутосомно-доминантные моногенные синдромы встречаются с частотой 1: 200 индивидов; заболевание наблюдается у многих поколений, передается потомкам и рецидивирует с частотой 50%. Примерами аутосомно-доминантных моногенных расстройств могут быть:
- ахондроплазия,
- нейрофиброматоз,
- синдром Марфана,
- болезнь Хантингтона,
- семейный полипоз.
Появление аутосомно-доминантных заболеваний у новорожденных от «здоровых» родителей может быть обусловлено следующими причинами:
1. Мозаицизм зародышевых клеток. Мутация может иметь место лишь в популяции зародышевых клеток. Итак, родители являются непораженными, но могут передавать мутацию потомкам.
2. Новые мутации. Рост возраста родителей ассоциируется с увеличением риска аутосомно-доминантных расстройств (ахондроплазии, танатофорной дисплазии, нейрофиброматоза, синдрома Аперта — краниосиностоз). Риск рецидивов у других детей не увеличивается.
3. Вариабельна экспрессия. Тяжесть заболевания может варьировать, и родители могут не распознать мягкие и субклинические мутации.
4. Уменьшенная пенетрантность. Родители могут иметь аномальный ген без клинических проявлений заболевания.
5. Неверное отцовство. Частота неверного отцовства достигает 15%.
Аутосомно-рецессивные моногенные заболевания проявляются в многочисленных родственников при наличии двух пораженных аллелей. Если оба родителя являются носителями пораженного гена, риск заболевания у потомства равен 25% при каждой беременности. Аутосомно-рецессивные заболевания включают кистозный фиброз, серповидно-клеточную анемию, фенилкетонурию, болезнь Тея-Сакса, Канавана и др.
При Х-сцепленных рецессивных синдромах (гемофилия и др.) мать-носитель пораженного гена передает его своим сыновьям. Итак, 50% сыновей могут быть больными и 50% дочерей будут носителями этого гена. Редкие Х-доминантные синдромы могут передаваться от каждого родителя каждому ребенку подобно аутосомно-доминантных синдромов. Фенотип может сильно варьировать, что связано со смешанной пенетрантностью, лионизацией (гетерохроматизацией) Х-хромосомы (синдром ломкой Х-хромосомы) и геномным импринтингом.
Экспансия тринуклеотидных повторов. Некоторые гены содержат участки тройных повторов (например, ССС). Такие участки являются нестабильными и могут увеличиваться в следующих генерациях, этот феномен получил название антиципации. Количество повторений определяет степень поражения индивида. Экспансия тринуклеотидных повторов составляет основу многочисленных генетических расстройств, таких как синдром ломкой (фрагильной) Х-хромосомы, миотоническая дистрофия и болезнь Хантингтона.
Синдром ломкой (фрагильной) Х-хромосомы является наиболее частой причиной семейной задержки умственного развития. Пораженные мужчины имеют типичные черты: большие уши, выступающая челюсть, большие яички, аутичное поведение, легкая или умеренная умственная отсталость. Женщины обычно менее поражены в связи с инактивацией Х-хромосомы.
Ген ломкой Х-хромосомы локализуется в Х-хромосоме и имеет три нуклеотидные повтора (ССС). Нормальные индивиды имеют 6-50 повторов, непораженные носители женского пола могут иметь 50-200 повторов, которые могут распространяться на мейоза до полной мутации при наличии более 200 повторов. Если имеет место полная мутация, ген инактивируется путем метилирования; плод будет пораженным. Тяжесть заболевания зависит от степени Х-инактивации у женщин, степени метилирования и мозаицизма размера повторов.
Женщины-носители премутации имеют 50%-й риск передачи гена с экспансией. Мужчины с премутациею фенотипически являются нормальными, но все их дочери будут носителями премутации. В случае трансмиссии мужчинам количество повторов остается стабильным. Тест на ломку Х-хромосому выполняется с целью выявления количества повторов и степени метилирования.
Что такое пренатальный скрининг
Пренатальный скрининг, диагностика и лечение является относительно новой проблемой в акушерстве. Началом пренатального скрининга была, возможно, эра ультразвуковой диагностики в акушерстве, которая началась около двух десятилетий назад. С открытием новых генов и их фенотипов становится все более возможным пренатальный генетический диагноз. Следует различать понятия скрининга и диагностики.
Пренатальный скрининг позволяет выявить индивидов высокого риска осложнений среди популяции индивидов с низким риском осложнений. Специфичность и чувствительность скрининговых тестов очень важны, учитывая возможность ложноположительных и ложноотрицательных результатов скрининга.
Пренатальная диагностика, конечно, более специфическая, чем скрининг (например, амниоцентез или биопсия хориона), но имеет и больший риск осложнений. Первым шагом по определению риска для плода является скрининг матери о наличии определенных состояний или заболеваний.
Нередко возникает вопрос о вероятности роста частоты врожденных пороков у потомков семейных пар, которые получали лечение по поводу бесплодия. Тяжелая олигоспермия и азооспермия ассоциируются со сбалансированными транслокациями хромосом (3-5%), синдромом Кляйнфельтера (47, ХХУ), аномалиями и микроделеции У-хромосомы.
Аномалии Х-хромосомы (ХХУ, ХХХ, Х-мозаицизм при синдроме Тернера) ассоциируются с пониженной фертильностью (субфертильностью), а также увеличением риска хромосомных аномалий у потомков. В 2/3 пациентов с врожденным отсутствием семявыносящих протоков имеет место хотя бы одна мутация гена, который отвечает за развитие кистозного фиброза. Итак, эти пациенты подлежат скринингу на наличие кистозного фиброза. Таким пациентам обычно показана интрацитоплазматическая инъекция сперматозоида в яйцеклетку, хотя наличие мутантного гена по кистозному фиброзу может влиять на репродуктивные намерения.
Заключение
1. Не каждую беременность удается сохранить на малых сроках. В большом проценте случаев выкидыши обусловлены хромосомными нарушениями у плода, и родить живого ребенка невозможно. Гормональное лечение может отсрочить момент выкидыша, но не может помочь зародышу выжить.
2. Повышенная нестабильность генома супругов является одним из причинных факторов бесплодия и невынашивания беременности. Выявить такие супружеские пары помогает цитогенетическое обследование с анализом на хромосомные аберрации. В некоторых случаях повышенной нестабильности генома специальная антимутагенная терапия может помочь повысить вероятность зачатия здорового ребенка. В других случаях рекомендуется донорская инсеминация или использование донорской яйцеклетки.
3. При невынашивании беременности, обусловленном хромосомными факторами, организм женщины может «запомнить» неблагоприятный иммунологический ответ на плодное яйцо (иммунологический импринтинг). В таких случаях возможно развитие реакции отторжения и на зародыши, зачатые после донорской инсеминации или с использованием донорской яйцеклетки. В таких случаях рекомендуется проведение специального иммунологического обследования.
Теги:
генетика, акушерство, гинекология