Самое дорогостоящее лекарство в мире
Содержание:
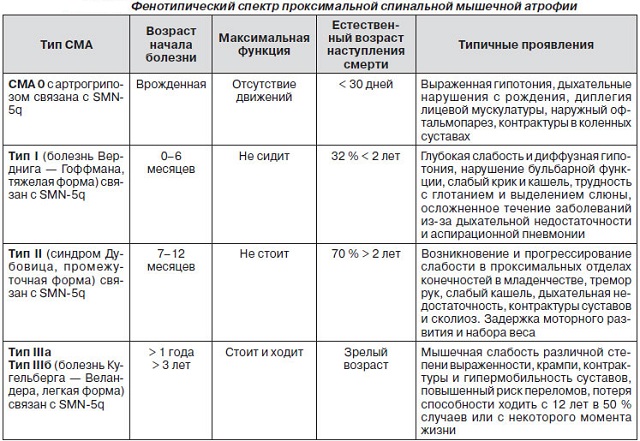
Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
СМА 0
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
СМА-1
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Поздняя младенческая
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.

Ювенильная
Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
Поздние патологии
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
«Врачи говорили — особенность развития»
Страшное генетическое заболевание было известно и раньше, но еще несколько лет назад специфического лечения СМА не существовало, и дети в лучшем случае оставались инвалидами, прикованными к креслам и аппаратам искусственной вентиляции легких. В худшем случае — умирали, не дожив и до пяти лет. СМА — это полное отсутствие или сложная поломка гена, отвечающего за работу двигательных нейронов. Ребенок сначала не может двигаться, потом — есть, затем — самостоятельно дышать. Мозг при этом остается полностью сохранным. Заболевание начинает проявляться в младенческом возрасте. Но, к сожалению, не все врачи даже столичных клиник готовы узнать СМА «в лицо».
Семья юриста Игоря и его супруги Анастасии, госслужащей, о существовании СМА, как и большинство счастливых российских семей, не подозревала. Дедушка маленького Гриши Зарина — бывший футболист и тренер футбольного клуба «Динамо-Брянск». Заботливая бабушка. Обычная российская семья.
Долгожданный сын Гриша немного отставал в развитии двигательной активности, но не критично, поэтому в апреле 2020 года, отправившись на обычный плановый осмотр, они не удивились предположению врача-ортопеда: гипотонус, особенность развития. Врач предложил массажи, сеансы физиотерапии, лечебную физкультуру. Но началась пандемия коронавируса, неподготовленным людям такие занятия проводить не приходилось, и они отправились в платный центр за консультацией. В частном центре врач оказался более внимательным, у него появились вопросы, и он посоветовал родителям обратиться к неврологу. Затем — хождения по врачам и инстанциям, генетический анализ удалось сдать только в июле. Ответ пришел 13 августа: СМА, предположительно, 2-го типа.
Семья до сих пор не может поверить в то, что услышала 13 августа. Но готова бороться за сына.

Федеральное агентство новостей  / 
Кариотипирование одного или обоих супругов
Кариотипирование — это исследование количественного набора хромосом, а также их структурных перестроек (хромосомных аберраций). Перестройки могут быть внутри- и межхромосомными, могут сопровождаться нарушением порядка фрагментов хромосом (делеции, дупликации, инверсии, транслокации). Хромосомные перестройки подразделяют на:
- Сбалансированные. Иинверсии, реципрокные транслокации не приводят к потере или добавлению генетического материала, поэтому их носители, как правило, фенотипически нормальны.
- Несбалансированные. Делеции и дупликации меняют дозовое соотношение генов, и, как правило, их носительство сопряжено с существенными отклонениями от нормы.
У здорового человека должно быть 22 пары аутосом и 1 пара половых хромосом (ХХ или ХУ). Для пар, страдающих от бесплодия, кариотипирование назначается скорее из-за перестраховки, его назначение более оправдано при привычном невынашивании, особенно если оно было связано с доказанной анеуплоидией эмбриона/ов.
У пациентов с бесплодием хромосомные перестройки встречаются редко. С привычным невынашиванием сбалансированные перестройки встречаются чаще, но занимают всего лишь 4–5% в структуре причин привычного невынашивания. При этом у таких пациентов всегда есть шанс на рождение здорового ребенка без проведения дорогостоящего обследования и лечения. Без соответствующих исследований и лечения риски повторного невынашивания и рождения ребенка с тяжелыми проявлениями несбалансированной транслокации существуют, но они достаточно низкие.
Так как анализ проводится на коммерческой основе, пара должна понимать, что выявление нарушений кариотипа повлечет за собой рекомендацию делать предимплантационную генетическую диагностику эмбрионов (ПГД) на конкретную хромосомную поломку, а также ПГТ-А для исключения численных хромосомных нарушений у эмбрионов.
Лечение спинальной мышечной атрофии
Ещё совсем недавно болезнь считалась неизлечимой и часто приводила к смерти или тяжёлой инвалидности. Все мероприятия были направлены на облегчение состояния пациента. В декабре 2016 года был разработан первый препарат Спинраза, который усиливает синтез белка SMN. Недостатком этого препарата является то, что он имеет высокую стоимость и лечение им нужно проводить на протяжении всей жизни больного СМА. Однако благодаря Спинразе были получены очень хорошие результаты в лечении и прогнозе у таких больных, особенно, если лечение начиналось на этапе доклинических проявлений. Дети в таком случае не отставали от своих здоровых сверстников по уровню физического развития.
В 2018 году был разработан препарат Золгенсма, который в мае 2019 года получил одобрение FDA. Он представляет собой генотерапевтическое лечение с заменой дефектного гена SMN1 на его нормальную копию. Переносчиком вектора AAV9 в клетку выступает инактивированный аденовирус. Итогом лечения становится нормальная выработка белка SMN. Человек получает полное исцеление после одной инъекции препарата. Дети, получившие Золгенсма на доклинической и ранней клинической стадии СМА, ничем не отличаются от своих здоровых сверстников. Даже пациенты, находящиеся на ИВЛ демонстрируют положительные результаты – начинают дышать самостоятельно, сидеть, вставать, ходить и т.д. Единственный недостаток препарата – это его стоимость. Он является самым дорогим на сегодняшний день препаратом в мире, 1 укол стоит более 2 млн.$. Активно ведётся разработка аналога Золгенсма в России и других странах мира, надеемся, что это произойдёт в ближайшем будущем, и многие семьи смогут получить спасительное лечение для своих детей.
Больным СМА также необходимо симптоматическое лечение по показаниям:
- метаболические препараты для улучшения функции митохондрий в мышечных тканях;
- противоэпилептические препараты – увеличивают образование белка SMN и улучшают клиническое течение болезни;
- муколитики и отхаркивающие – ввиду слабости дыхательной мускулатуры в лёгких скапливается мокрота, облегчить выведение которой помогут данные препараты;
- прокинетики и ИПП – в связи с нарушением глотания и снижением активности ЖКТ, а также частых ГЭРБ у больных СМА;
- гормональные препараты – при развитии сопутствующих осложнений;
- сахароснижающие лекарственные средства – в случае развития сахарного диабета на фоне СМА.
При развитии крайне выраженных контрактур суставов, деформаций грудной клетки и позвоночника рекомендованы ортопедические операции. Больным с постоянными рецидивами пневмоний ввиду аспирации пищи требуется проведение трахеостомии.
Месяц осведомлённости о спинальной мышечной атрофии


августемесяц осведомленности о СМА (спинальной мышечной атрофии)фонд «Семьи СМА»http://f-sma.ru/205.html
С помощью ссылки http://twibbon.com/support/месяц-осведомленности-о-сма можно добавить эмблему акции на фотографию своего профиля в Twitter или Facebook, или просто сохранить фото и использовать на любом другом ресурсе.
Разместите у себя на страничке в социальной сети фото с эмблемой акции на срок, который будет для Вас оптимальным. Так вы поможете рассказать об этом нервно-мышечном генетическом заболевании. Вместе мы можем сделать больше!
Техническая подсказка: приложение создает альбом Twibbon Photos в разделе «Ваши фотографии» на Фейсбук, если захотите установить фото с эмблемой в качестве фотографии профиля, откройте фото и в левом нижнем углу нажмите «сделать фото профиля». Или сохраните изображение на свой компьютер и сможете ставить картинку профиля на любом другом ресурсе.
Дифференциальный анализ
Прогнозирование возможно благодаря полученным данным в результате:
- генетического анализа;
- особенностей клиники (тип болезни, место локализации);
- сопутствующие симптомы, к примеру, фасцикуляция языка;
- результатах ЭКГ и данных биопсии скелетных мышц.
Дифференцировать младенческий и ранний вид СМА можно на основании проблем с врожденной мышечной гипотонией – синдрома «вялого ребенка», амиатонии, врожденного характера доброкачественной дистрофии мышц, наследственности и хромосомного анализа.
Юношеский вид отличается от спинальной амиатрофии Кугельберга-Вееландер и разных видов мышечных дистрофий.
Денис Хайруллин, папа Варвары
— Мы познакомились с супругой 12 лет назад, оба мы из Благовещенска, сейчас живём в Уфе. Я работаю в сфере сотовой связи: начинал с позиции, теперь я руководитель розничной сети, у меня в подчинении 25 магазинов. Жена работала администратором, потом ушла в декрет. У нас есть старшая дочка Ева, ей 4,5 года. А 12 февраля 2019 года на свет появилась Варя.
Когда младшей дочке было девять месяцев, мы забили тревогу. В таком возрасте ребёнок должен уже вставать, а некоторые уже ходят. Варя не могла встать, ножки были очень слабые, так что мы начали ездить по больницам. Только летом 2020 года нас направили сдавать генетический анализ, и лишь в сентябре поставили диагноз. Мы потеряли много времени.
Когда мы начали узнавать что-то о СМА, то погрузились в кошмар, из которого, казалось, нельзя выбраться. Но мы сразу решили, что лечиться будем «Золгенсмой»: препарат вводится только один раз, к тому же два других лекарства, «Спинразу» и «Эврисди», у нас в регионе всё равно быстро не достать.
Сбор мы открыли 28 сентября 2020 года. Информацию о Варе помогали распространять родственники, друзья, потом присоединились незнакомые люди из Уфы и Благовещенска. По согласованию с администрациями этих городов мы провели автопробег, благотворительные ярмарки при поддержке спонсоров. Волонтёры в соцсетях пытались достучаться до российских звёзд, известных блогеров, писали в различные компании. Конечно, было много отказов, но неравнодушные люди всё равно не сдавались.
За два с половиной месяца нам удалось собрать чуть больше 10 млн рублей, этого мало для закупки лекарства. Параллельно мы собирали необходимые документы, чтобы подать заявку на участие в лотерее, которую устраивает производитель «Золгенсмы». Мы использовали все свои возможности. Где-то что-то должно было выстрелить.
С сентября у нас с женой не было ни одного выходного дня, мы физически очень устали, у нас почти опустились руки. И вот поздно вечером 10 января 2021 года супруге позвонила невролог Вари и, плача от радости, сообщила, что мы выиграли в лотерею.
Представьте, что у тебя на плечах был огромный камень, ты уже выбился из сил его тащить, уже готов упасть и не встать. А потом в один момент он у тебя слетает. Вот что мы почувствовали тогда.
Сразу после того, как мы узнали о выигрыше, объявили о закрытии сбора. Из собранных денег мы оставили 3 млн на дальнейшую реабилитацию и закупку вертикализатора. 6,9 млн мы перевели на счёт маленького «смайлика» Саши Горобинского. Около 442 тыс., собранных через второй наш фонд, отдали Матвею Куликовскому, «смайлику» из Санкт-Петербурга. Порядка 100 тыс. рублей, которые собрали на зарубежной площадке, перевели мальчику с трахеостомой из Уфы, а деньги, которые поступили на счёт после закрытия сбора, — это около 20 тыс. рублей — перевели девочке-«смайлику» из Барнаула.
7 февраля мы, отсидев карантин, чтобы Варя не заболела накануне госпитализации, прилетели в Москву. 11 февраля, уже после сдачи всех анализов, нашей дочке сделали инъекцию «Золгенсмы» в НИКИ педиатрии им. Вельтищева. На следующий день Варе исполнилось два года.

Дочка ещё слабенькая, изменения пока трудно заметить — слишком мало времени прошло. Через месяц показатели печени должны прийти в норму, и тогда будем смотреть на эффект.
Мы с женой понимаем, что впереди ещё много работы, ведь многое зависит от реабилитации. Помимо вертикализатора, купим инвалидное кресло, хотя сразу решили, что не будем к нему именно как к инвалидному относиться — это просто средство, чтобы Варя передвигалась куда захочет. Плюс она пройдёт месячную реабилитацию в центре нашего фонда «Мать и дитя».
Наша старшая дочь Ева раньше ходила в садик, но после того, как мы узнали диагноз Вари, пришлось забрать её оттуда. Детям со СМА противопоказано любое заболевание, а в садиках, сами знаете, малыши часто болеют. Поэтому сейчас Еву учит моя мама, она преподаватель русского языка и литературы.
Варя улыбчивая, позитивная и подвижная. Даже врачи говорят, что для такого диагноза наш ребёнок прямо живчик. И мы очень надеемся увидеть эффект от лекарства через месяц.
Генетические факторы болезни
Спинальная мышечная атрофия появляется при наследовании рецессивного генома 5 хромосомы. Если оба человека, которые родили младенца, являются носителями СМА, то с вероятностью не менее 25% передают ген ребенку. В результате нарушается синтез белковых структур, разрушение моторных нейронов спинного мозга происходит в несколько раз быстрее, чем восстановление.
В период эмбрионального развития нервная система ребенка производит только половину от необходимого объема моторных нейронов. Со временем при СМА этот процесс сильно замедляется. После рождения из-за недостатка структур развивается спинальная атрофия.

Особенности функционирования нейронов
Активный мозг постоянно посылает импульсы в спинной мозг, а проводниками служат нервные клетки. Они доставляют сигналы в мышцы, в результате чего запускается их движение. Если этот процесс нарушен, то движение становится невозможным.
При спинально-мышечной атрофии двигательные нейроны ног, входящие в состав спинного мозга, работают некорректно. Они отвечают за сигналы, с помощью которых мозг поддерживает такие функции, как ползанье, поддержка шеи, сжимание и движение руками, ногами, а также дыхание и глотательный рефлекс.
В результате мышцы, не получающие постоянных сигналов, начинают атрофироваться.
Симптомы разных форм болезни
Существует общий набор признаков СМА, по которым можно заподозрить патологию, если других проблем не обнаруживается, либо диагноз вызывает сомнение. Группу симптомов сводят к проявлению вялого периферического паралича:
- выраженная мышечная слабость или атрофия разных мышечных групп;
- сначала в процесс вовлекаются конечности – симметрично, ноги, а затем руки, постепенно втягивается и туловища;
- отсутствуют расстройства чувствительности и тазовые нарушения;
- наиболее выраженные проблемы затрагивают проксимальные или дистальные мышечные группы.
У пациентов появляются подергивания и фибрилляции – мерцательные аритмии.
Признаки СМА1
Заболевание Верднига-Гоффмана бывает 3 видов:
- Врожденная форма. Начинается в течение 1-6 месяца жизни, обладает тяжелыми симптомами. Обнаружить признаки можно во внутриутробном развитии – эмбрион будет мало двигаться. Гипотония наблюдается сразу же после рождения ребенка. Такие младенцы не держат голову, не могут сидеть. Постоянно находятся в позе лягушки с раздвинутыми конечностями. Сначала симптомы появляются в ногах, затем в руках, после этого страдает дыхательная мускулатура. Психическое развитие у таких детей медленное, они редко доживают до 2 лет.
- Ранняя спинальная мышечная атрофия. Первые признаки начинают беспокоить пациента до 1,5 лет, чаще всего – после какой-либо инфекции. Даже если раньше ребенок мог стоять и сидеть, теперь он утрачивает эти функции. Развиваются парезы, а затем поражаются дыхательные мышцы. Ребенок умирает, как правило, в результате затяжной пневмонии или дыхательной недостаточности в возрасте 3-5 лет.
- Поздняя форма. Патология встречается после 1,5 лет, двигательные возможности сохраняются у ребенка до 10 лет. Медленный прогресс симптомов приводит к дыхательной недостаточности и смерти в возрасте до 18 лет.
Признаки болезни Кугельберга-Веландера
Возникает в возрасте от 2 до 15 лет. Сначала в процесс вовлекаются нижние конечности, затем пояс таза, на последних стадиях страдает плечевой пояс и дыхательная система. Примерно у 25% пациентов появляется синдром псевдогипертрофии мышц, из-за чего патологию путают с мышечной болезнью Беккера.
Спинальная мышечная атрофия Кугельберга-Веландера не сопровождается костными деформациями, и пациенты способны сами себя обслуживать в течение долгих лет.
Амиотрофия Кеннеди
Эта патология входит во взрослую группу, болеют представители мужского пола после 30 лет. Женщины от патологии не страдают. Течение – умеренное, сначала поражаются ножные мышцы, последующие 10-20 лет пациент сохраняет привычный ритм жизни. Только после этого начинают страдать мускулы рук и головы. У многих пациентов со временем наступают эндокринные изменения: атрофия яичек, отсутствие либидо, сахарный диабет.
Дистальная СМА
Эта форма спинально-мышечной атрофии также развивается у взрослых пациентов после 20 лет. Ее второе название – СМА Дюшенна-Арана. Риск развития патологии сохраняется вплоть до 50 лет. Атрофия начинается в руках, вызывает синдром «когтистой лапы», потом переходит на крупные мышцы. Со временем появляются парезы мышц нижних конечностей, а туловище страдает редко. Прогноз у этой формы благоприятный, если не присоединяется торсионная дистония или болезнь Паркинсона.
СМА Вюльпиана
Скапуло-перонеальная форма спинально-мышечной атрофии, сопровождаемая симптомом «крылатых» лопаток. Появляется в среднем в 20-40 лет, позже встречается реже. Поражается плечевой пояс, а через некоторое время – руки и нижние конечности. При этой форме спинальной болезни двигательные функции у пациента сохраняются на 30-40 лет.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс
Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
1.Общие сведения
В современной неврологии одной из острых и актуальных проблем, для которой существуют пока лишь паллиативные решения, остается группа спинальных мышечных атрофий (СМА, SMA). Речь идет о наследственных заболеваниях, при которых из-за дефекта одного из генов изначально нарушен механизм питания т.н. моторных нейронов спинного мозга – нервных клеток, отвечающих за сократительную активность мускульных структур организма, – что приводит к постепенному их отмиранию.
К счастью, такая патология достаточно редка, однако среди наследственных заболеваний она является одной из наиболее часто встречаемых. Статистические данные по спинальной мышечной атрофии в различных источниках варьируют (примерно один случай на 6-8 тыс). Достоверно известно, что вероятность рождения больного ребенка при носительстве сбойного гена обоими родителями составляет 1/4 (25%); установлено также, что в случае «запуска» атрофического процесса с самого рождения шансы дожить до двухлетнего возраста не превышают 50%. Однако СМА может начаться в любом возрасте, обычно в интервале 20-50 лет.
Выделяют четыре типа спинальной мышечной атрофии. Синдром Кеннеди (спинобульбарная мышечная атрофия, СБМА) представляет собой взрослую смешанную форму, имеющую ряд отличительных особенностей. В частности, СБМА чаще встречается и протекает тяжелее у мужчин, чем у женщин; это связано с тем, что дефектный ген находится в Х-хромосоме.
Суды и сборы
По данным руководителя фонда «Семьи СМА» Ольги Германенко, по всему миру лечение «Спинразой» получили 10 тыс. человек. В России по состоянию на середину декабря начали или продолжили инъекции этого препарата 209 человек, из них восемь взрослых.
«Показатель хороший, но недостаточный», — констатирует она.
При этом, по словам Ольги Германенко, именно в 2020 году некоторые семьи пациентов со СМА столкнулись с затягиванием процесса назначения препарата, в том числе и через суд. Среди доводов региональных властей — неопределённость источников и порядка финансирования закупок, отсутствие «Спинразы» в перечне жизненно необходимых и важных лекарственных препаратов (ЖНВЛП), а также малая эффективность терапии.
24 ноября 2020 года «Спинраза» была внесена в перечень ЖНВЛП. Теперь государство может контролировать стоимость лекарств в перечне, а региональные минздравы лишились возможности отказывать в предоставлении дорогостоящего препарата под предлогом его отсутствия в списке.
Кроме того, производитель «Спинразы» выразил готовность снизить её цену на 25%, таким образом, каждый четвёртый пациент сможет лечиться за счёт этой экономии. Также на основании перечня ЖНВЛП формируются так называемые территориальные программы медицинской помощи — значит, регионам будет проще закладывать соответствующий бюджет.
Через два дня после включения «Спинразы» в перечень жизненно необходимых лекарств произошло ещё одно важное для всех пациентов со СМА событие: в России был зарегистрирован препарат «Эврисди». К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке
К тому же появление второго зарегистрированного препарата в перспективе может ещё снизить цены на рынке.
Регистрация в России самого дорогого лекарства от СМА, «Золгенсмы», ожидается в 2021 году. Пока этого не произошло, семьи пациентов либо собирают деньги на лечение в частном порядке (единичные сборы даже удалось закрыть), либо судятся за право получить терапию за счёт государства.
Особенность «Золгенсмы» в том, что, согласно инструкции, препарат надо применить до того, как ребёнку исполнится два года, тогда как терапию «Спинразой» и «Эврисди» можно начать в любом возрасте.
Опираясь именно на этот довод, суд отказал в предоставлении «Золгенсмы» Косте Гепалову из Санкт-Петербурга, которому в сентябре исполнилось два года. Однако летом специалисты в ЕС, а с ними и американские медики пришли к выводу, что важнее критерий веса ребёнка, а не его возраста. Семья Гепаловых подала повторный иск в суд с новыми документами и ожидает решения.
Общая информация
- СМА — одна из наиболее частых причин детской смертности, вызванной наследственными заболеваниями.
- Спинальные мышечные атрофии детского возраста наследуются по аутосомно-рецессивному типу.
- Ген спинальной мышечной атрофии картирован на 5-й хромосоме, q11.2 — 13.3.
- Ген СМА был идентифицирован в г., его обозначение SMN (survival motor neuron).
- В среднем один из 6000 — 10000 детей рождается со СМА, в разных странах частота сильно различается.
- 50 % детей с СМА не доживают до двух лет (это дети преимущественно с 1-й формой заболевания).
- SMA может проявиться в любом возрасте, «мягкие» формы проявляются в среднем и пожилом возрасте.
- В среднем каждый 50-й человек имеет рецессивный ген, способный вызывать СМА.
- В соответствии с менделевским расщеплением ребёнок двух носителей поражается СМА с вероятностью 25 %. В этом случае оба родителя несут одиночный дефектный ген, но защищены присутствием второго, нормального гена, который является вообще достаточным для нормальной функции организма. Две дефектных копии гена приводят к генному нарушению, так как не обеспечивается синтез необходимого белка.
- В ходе медико-генетического обследования нескольких российских и среднеазиатских популяций (1,8 млн человек) выявлено 33 больных спинальной мышечной атрофией (СМА): 29 с детской проксимальной СМА (СМА I—III) и 4 c редкими формами. Выявлено «перекрывание» проявлений разных типов СМА I—III (I—II и II—III) у части больных, внутрисемейные различия типов в 3 из 6 семейных случаев, клинико-генетический полиморфизм редких форм СМА. (Г. Е. Руденская, Р. А. Мамедова).
Мы с мужем были как загнанные звери
Днем и ночью мы молились, чтобы Леша смог задышать без аппарата ИВЛ, и мы забрали бы его из больницы домой.
В один из дней мы приехали навестить Лешу в реанимации. К нам вышла заведующая и сказала: «Алексея здесь нет. Его перевезли в другую реанимацию». Я спросила: «Как перевезли? У него ведь даже одежды не было». Заведующая говорила, чтобы я не переживала, что такие дети могут жить долго, до 18 лет, что Леша хорошенький ребенок… Я не слушала, что она говорит, у меня в голове застряли только ее слова: «Сейчас главное здоровье вашего Алексея». Что она хотела сказать?
Мы сразу поехали в больницу в Измайлово, куда перевели Лешу. И пожалели, что раньше не соглашались туда перевестись. Там маленькая реанимация, всего 3 ребенка, родителям можно находиться целый день. Местные реаниматологи продолжали настаивать на трахеостоме. Одна врач уговаривала нас: «Почему вы не соглашаетесь? Он с трахеостомой даже говорить сможет научиться. Он еще долго проживет. Все будет хорошо». Но мы понимали, что это неправда, у Леши была самая агрессивная форма СМА, с которой дети не живут долго…
Целыми днями мы проводили с Лешей в реанимации. Из-за трубок я не могла взять его на ручки, покачать, не могла дать соску. Я просто сидела рядом и гладила его по голове.
Когда я была рядом, он почти не плакал, спокойно засыпал. Я понимала, что потом буду очень жалеть о том месяце в предыдущей реанимации, когда я не могла с ним проводить время. Реаниматологи стали тренировать Лешу дышать самостоятельно. Потом из хосписа нам привезли аппарат для неинвазивной вентиляции, чтобы попробовать перевести Лешу с трубки на маску.
Однажды я заметила, что у Леши необычно много слизи в горле, позвала дежурного реаниматолога. Реаниматолог пришла очень злая и сказала: «Я же вам говорила, что надо ставить трахеостому. У него сместилась интубационная трубка, и теперь так будет повторяться очень часто».
Она унесла Лешу и вернула мне его через 15 минут, всего окровавленного. У Леши были бешеные глаза. Раз он не спит, значит, ему не давали наркоз, когда переставляли трубку? Я спросила: «Вы что, не давали ему наркоз?». Реаниматолог ответила: «А зачем ему наркоз, у него же СМА, у него нет рефлексов». Но ведь он все чувствует, ему было больно!!!
Я поняла, что нужно выписываться домой как можно скорее, иначе этот ужас может повториться. Просила нашего врача поскорее попытаться перевести Лешу на вентиляцию через маску. И это удалось сделать. Леша дышал на маске спокойно, поулыбался и быстро заснул. Заведующая паллиативным отделением сказала, что завтра в 9 утра мы можем выписываться. Но какое-то чувство внутри подсказывало мне, что надо ехать домой сегодня, иначе ситуация, когда Леше без наркоза вставляют в горло трубку, может повториться снова.
Заведующая отделением не хотела отпускать нас вечером. Она кричала и сказала, что обратится в полицию, если мы уедем вечером из больницы.
И действительно, мне на мобильный позвонили с незнакомого номера: «Здравствуйте, это отдел по делам несовершеннолетних. Где вы сейчас находитесь?
Вы действительно хотите забрать ребенка из больницы против воли врачей?». Сказали, что будут «держать меня на контроле», чтобы я не причинила вред своему ребенку.
Мы с Лешей еще были в больнице, а к нам домой приехала полиция на 2 машинах. Они звонили в дверь, но муж не открыл.
Я понимала, что если мы сейчас останемся в больнице, Леша может снова попасть на аппарат ИВЛ, в реанимацию, и другого шанса забрать его домой у нас уже больше может не быть. Но я не знала, что делать. И тут уже поздно вечером мне позвонил главный врач этой больницы и сказал: «Вы можете смело ехать домой. Это безобразие, что врачи вызвали полицию. Вот мой мобильный телефон, при любых проблемах звоните мне хоть ночью. Полиция уже уехала».
Я вызвала частную машину скорой помощи, и мы поехали домой. Леша улыбался, мне кажется, он понимал, что мы едем домой.
Дома мы каждые 3 часа кормили его через зонд, откашливали с помощью откашливателя, санировали. Леша дышал с помощью аппарата неинвазивной вентиляций легких через маску. Мы с мужем брали его на ручки, качали, разговаривали с ним, читали вслух.
На следующий день к нам домой пришла инспектор по делам несовершеннолетних. Очень долго задавала нам с мужем вопросы, протоколировала ответы. Какой у ребенка диагноз, когда мы попали в больницу, когда выписались, как он себя чувствует.
Лёша. Фото: facebook.com/lida.moniava
Осложнения ишемического инсульта
Возникающие осложнения при инсульте определены тяжёлым состоянием больного и ограниченной его возможностью к самостоятельному обслуживанию и передвижению.
Возможные осложнениями ишемического инсульта:
- Тромбоэмболия лёгочной артерии — тяжелейшее осложнение инсульта. С целью профилактики, больному надевают ортопедические компрессионные чулки или применяют специальные устройства для пневмокомпресси ног.
- Пневмония. Профилактика этого осложнения направлена на поддержание свободного состояния верхних дыхательных путей, уход за полостью рта, поворачивание больного каждые два часа во избежание застоя в лёгких, своевременное назначение антибиотиков.
- Пролежни — серьёзная проблема для больных, переносящих инсульт. Профилактику пролежней необходимо начинать с первых дней заболевания. Для этого необходимо следить за чистотой белья, устранять складки на постельном белье, обрабатывать тело комфортным спиртом, присыпать тальком складки кожи, подкладывать круги под крестец и пятки. Профилактика пролежней требует поворота больного с интервалом не реже 2-3 часов.
- Контрактура — ограничение движения в суставе. Профилактику контрактур начинают при первой возможности, выполняя пассивные движения парализованными конечностями. Во избежание развития мышечных контрактур при наличии гемипареза или гемиплегии парализованные конечности укладывают в положении, противоположном обычной позе Вернике-Манна .
- Потеря памяти или проблемы с мышлением. Многие люди, перенесшие инсульт, испытывают потерю памяти, у других возникают трудности с мышлением, проблемы с речью, в том числе с её пониманием, чтением или письмом.
- Эмоциональные проблемы. Людям, перенесшим инсульт, труднее контролировать свои эмоции или у них может развиться депрессия и изменения в поведении .
Также необходимо следить за мочеиспусканием, вовремя катетеризировать мочевой пузырь. В случае запора назначают клизму.
При стабилизации общего состояния проводят пассивную гимнастику, общий массаж мышц. По мере стабилизации переходят к обучению больных сидению, самостоятельному стоянию, ходьбе и навыкам самообслуживания.