Синдром ретта: как бороться с генетической ошибкой?
Содержание:
Классификация болезни
Единственное разделение такой болезни у малышей предполагает наличие нескольких этапов прогрессирования, в зависимости от которых будет отличаться клиническая картина:
1 стадия — начинается с момента достижения ребенком 4 месяцев вплоть до 2-летнего возраста
В таких случаях начинают слабо выражаться первые признаки болезни, отчего родителям очень важно следить за поведением младенца. Лечение, начатое на таком этапе, может остановить прогрессирование синдрома Ретта;
2 стадия — отличается прогрессированием, а клиницисты диагностируют ее как энцефалит
Может манифестировать в возрастной категории от 1 года до 3 лет;
3 стадия — охватывает весь дошкольный период и ранний школьный возраст. Отличается стабильным протеканием и возможным улучшением состояния ребенка;
4 стадия — проявляется примерно к 10 годам. Выражается в обездвиживании детей и формировании вторичных костных деформаций.
История наблюдательного педиатра
В феврале 1965 года педиатр из Австрии Андреас Ретт открыл болезнь, которая позднее получила его имя. Сам он так описывает этот эпизод: «В приемной врача сидели двое матерей, которые держали своих дочерей на коленях. Девочки раскачивались из стороны в сторону, а женщины держали их за руки. Вдруг девочкам удалось освободить руки и они, словно по команде, стали совершать идентичные характерные «моющие» движения. На их лицах застыло одинаковое выражение, их руки двигались по одним и тем же траекториям, взгляд одинаково был направлен вперед. Я попросил матерей не останавливать их, и меня озарила идея…»
В следующем году Ретт объездил Европу в поисках аналогичных случаев «синдрома атрофии мозга», как он назвал обнаруженную им патологию. Долгое время болезнь считали то разновидностью аутизма, то ранней злокачественной шизофренией. Только в 1983 году болезнь была признана как «отдельная нозологическая единица» и получила свое нынешнее название. А в 1984 году уже появилась Международная ассоциация синдрома Ретта.
Данная патология — не из самых редких заболеваний. Оно развивается у одной из 10 тысяч или даже из 15 тысяч девочек, в отдельных регионах мира этот показатель составляет даже 1:3000. Сегодня болезнь Ретта считается одной из самых частых причин умственной отсталости у девочек.
Что представляет собой синдром Ретта?
Синдром Ретта – психоневрологическая болезнь генетического происхождения, которая проявляется в виде тяжелой формы умственной отсталости. Данное заболевание вызывает остановку развития ребенка в детском возрасте, провоцируя регресс эмоциональных, интеллектуальных и физических достижений, а также становится причиной множественных нарушений здоровья.
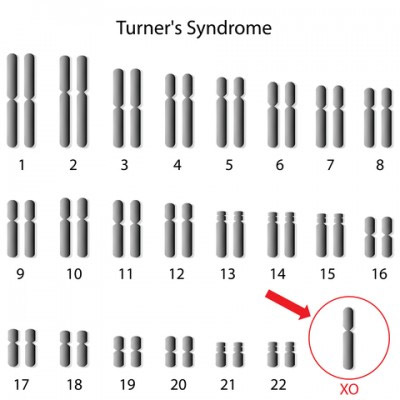
Развитие патологии основывается на генных мутациях, связанных с поражением Х-хромосомы. Так как у девочек Х-хромосома парная, то и заболевание диагностируется исключительно у них. Мальчики имеют лишь одну Х-хромосому, поражение которой не совместимо с жизнью. Патология возникает у одной девочки из десяти-пятнадцати тысяч и является одним из наиболее распространенных заболеваний среди всех наследственных видов умственных отклонений.
Дети с синдромом Ретта нуждаются в активной работе со специалистами, постоянной реабилитации и восстановлении. Только так возможно максимально сохранить двигательные и умственные навыки ребенка.
К каким докторам следует обращаться если у Вас Синдром Ретта:
Психиатр
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Ретта, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Патогенез
Науке известно 800 различных мелких мутаций, приводящих к синдрому. Если ген (МEСP2), находящийся на плече Х-хромосомы дефектен, то это приводит к сбою.
Ген МЕСР2, где происходит мутация, отвечает за кодирование метил-СpG-связывающего белка 2 (МеCр2). Больше всего этого белка содержится в нейронах (нервных клетках). Однако на начальных этапах формирования эмбриона этого белка мало, по мере взросления он увеличивается, а значит, прогрессирует мутация. Если ген не поврежден, то на определенном этапе его белок отключает некоторые другие гены, а развитие мозговых структур происходит нормально. При мутации же никакого отключения нет, а мозг развивается неправильно.
Новорожденный выглядит нормально, идет обычное правильное формирование, но ближе к 6-18-месячному возрасту начинается частичный или полный регресс психического развития и приобретенных навыков. Наблюдается аномалия походки, присоединяются дыхательные расстройства, судороги, речевые проблемы.
Размеры мозга у пациентов с синдромом маленькие, имеется атрофия коры и мозжечка, но разрушения нейронов нет. Это не типичная нейродегенеративная болезнь. Нейроны меньше по размеру, чем должны быть, они более плотно размещены, а ветвление дендритов (отростки от тел нейронов) упрощенное.
Популярные советы
- Что меняется в каждом знаке Черты, характерные для каждого знака…
- На эти 6 знаков зодиака Ожидается ли в апреле снегопад?…
- Гороскоп психических отклонений по знакам Психиатры считают, что не бывает…
- Щедрые и жадные мужчины по Иногда уже на первом свидании…
- Лучшие и худшие партнеры для Люди выбирают себе спутника жизни…
- 7 лучших мужей по знаку Описание и черты «лучшего» мужа…
- Как знаки зодиака относятся к Измена может перечеркнуть и разрушить…
- Тест: насколько сильно вы влюблены? Вы влюблены по уши в…
- Самые сильные стороны знаков зодиака Суперсила есть в каждом из…
- Кого ждет удача в любви Год Белого Быка принесет кардинальные…
Диагностика Синдрома Ретта:
Диагностические критерии синдрома Ретта по Е. Trevathan
Обязательные:
- 1) нормальный пре- и перинатальный периоды;
- 2) нормальное психомоторное развитие в течение первых 6- 18 мес. жизни;
- 3) нормальная окружность головы при рождении;
- 4) замедление роста головы в период от 5 мес. до 4 лет;
- 5) потеря приобретенных движений рук в возрасте от 6 до 30 мес., связанная по времени с нарушением общения;
- 6) глубокое повреждение экспрессивной и импрессивной речи, грубая задержка психомоторного развития;
- 7) стереотипные движения рук, напоминающие выжимание, стискивание рук, хлопки, «мытье рук», потирание их, появляющееся после потери целенаправленных движений;
- 8) появление нарушений походки (апраксии и атаксии) в возрасте 1-4 лет.
Дополнительные:
- 1) дыхательные расстройства (периодическое апное во время бодрствования, перемежающееся гипервентиляцией, аэрофагия);
- 2) судорожные припадки;
- 3) спастичность, часто сочетающаяся с дистонией и атрофией мышц;
- 4) сколиоз;
- 5) задержка роста;
- 6) гипотрофичные маленькие ступни;
- 7) ЭЭГ аномалии (медленный фоновый ритм и периодическое замедление ритма до 3-5 Гц, описаны центральновисочные Spikes как при фрагильной Х-хромосоме и роландической эпилепсии).
Дифференциальная диагностика
Дифференциальный диагноз синдрома Ретта и раннего детского аутизма (РДА) (Международная конференция по синдрому Ретта, 1988)
|
Признак |
Cиндром Ретта |
РДА |
|
Аутистические черты в возрасте 6-18 мес. |
Отсутствуют |
Часто проявляются |
|
Стереотипии |
Ритмические движения обеих рук по средней линии тела |
Более сложные и не по средней линии |
|
Стереотипные манипуляции с предметами |
Отсутствуют |
Типичны |
|
Моторика туловища и походка |
Прогрессирующие атаксия и апраксия |
Манерность, иногда грациозность позы и походки |
|
Судорожные припадки |
Значительно большие частота и полиморфизм |
Значительно меньшие частота и полиморфизм |
|
Расстройства дыхания, бруксизм, замедление роста головы и конечностей |
Типичны |
Отсутствуют |
Диагностика болезни
Перед постановкой диагноза врач изучает историю его болезни, беседует с родственниками, проводит собственные наблюдения. Для выявления характерных признаков болезни и исключения других заболеваний, имеющих схожую картину, применяются аппаратные методики, в том числе, компьютерная и магнитно-резонансная томография.
На ранних стадиях врачи для диагностики используются различные нейропсихологические тесты, поскольку неврологический осмотр, как правило, отклонений не выявляет. Поэтому исследование должно быть более обширным. При оценке состояния больного проводится беседа с его родственниками, поскольку сам пациент не способен заметить нарушений и больным себя не считает.
По результатам анализа крови выявляются причины деменции, которые не связаны с прогрессированием болезни Альцгеймера. Устранение их может способствовать тому, что симптомы заболевания будут устранены.
Сравнительно новыми способами диагностики заболевания являются методики, позволяющие наблюдать бета-амилоидные скопления в тканях головного мозга при помощи ПЭТ-сканера. О развитии заболевания также можно судить по количеству тау-белка или бета-амилоида в биологической жидкости из спинного мозга пациента.
Как правило, диагностика на ранних этапах болезни Альцгеймера проводится редко. Прежде всего, это связано с тем, что ни сам больной, ни его родственники не замечают отклонений от нормы и не обращаются за квалифицированной помощью. Диагноз ставится уже тогда, когда функциональные нарушения становятся слишком явными.
Как уже отмечалось выше, пациенты, в конечном итоге, погибают не от самой болезни Альцгеймера, а от сопутствующих заболеваний. Продолжительность жизни больного зависит от разных факторов, но исследователи отмечают, что в среднем женщины живут дольше мужчин.
К каким докторам следует обращаться если у Вас Синдром Ретта:
Психиатр
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Ретта, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Лечение
Лечение синдрома Ретта основано на симптоматическом лечении.
Устранить причину развития болезни и дальнейшее ее прогрессирования невозможно, но можно его замедлить.
Лечение синдрома Ретта включает в себя: медикаментозную терапию, профилактическую и лечебно-физическую культуру.
- С лечебной целью для купирования эпилептиморфных припадков используют антиконвульсанты (Дифенин, Диазепам, Клоназепам, Карбамазепин).
- Для регулирования биологических ритмов (день/ночь) используют Мелатонин.
Профилактические мероприятия направлены на устранение венозного застоя в сосудах малого и большого кругов кровообращения, предупреждения развития острой сердечной недостаточности, развития отека легких, инсульта, субарахноидальных кровоизлияний.
- Для улучшения мозгового кровообращения и кровообращения сосудов всех органов и систем используют: Фелодипин, Сермион, Инстенон.
- Для предотвращения тромбообразований используют антиагреганты (Аспирин) и антикоагулянты (Гепарин).
- Для улучшения мозговой деятельности используют ноотропные препараты: Пирацетам, Ноотропил, Пантогам, Актовегин.
- Средства для стабильной работы органов брюшной полости: сердца, печени и др.
- Используются гомеопатические средства для борьбы с умственной отсталостью (только по назначению врача).
Реабилитационные мероприятия при синдроме Ретта.
Прием лекарственных средств необходимо сочетать с лечебной физической культурой (ЛФК) и другими методами реабилитации.
ЛФК позволяет улучшить осанку ребенка и предупредить развитие искривления позвоночника или прогрессирования сколиоза.
С помощью ЛФК можно научить ребенка передвегаться. ЛФК необходимо сочетать с приемами мануального терапевта и лечебным массажем.
Наиболее эффективна при синдроме Ретта музыкотерапия по схеме Томатиса. Специально созданная музыка, прослушиваемая детьми, ведет к улучшению психоэмоциональных показателей ребенка.
Они начинают вступать в контакт с детьми и взрослыми. Положительной стороной является то, что музыкотерапию можно проводить в домашних условиях.
Для того, чтобы ребенок адаптировался к окружающей среде можно использовать:
- арт-терапию: творческие занятия в группе или отдельно в домашних условиях музыкой, рисованием и др.;
- иппотерапию: верховые прогулки на лошади;
- гидрореабилитацию: лечение «водой», например: минеральные ванны, гидромассаж и др.;
- дельфинотерапию: плавание в бассейнах с дельфинами;
- специальные занятия с собаками/кошками.
Также необходимы консультации логопедов, психологов, которые способны помочь ребенку «обрести себя», идти на контакты со взрослыми, выходить из депрессивного синдрома.
Питание
При данной патологии должно быть соблюдено лечебное питание.
Диета с повышенным содержанием жиров + витамины и клетчатка.
Необходимо кормить ребенка высококалорийными продуктами и специальными веществами для увеличения массы тела. Прием пищи должен быть каждые 3 часа.
В случае недоедания развивается кахексия (резкое снижение веса).
Профилактика и прогноз
Специально предназначенных профилактических мер, предупреждающих развитие недуга, не существует, поскольку неизвестны причины синдрома Ретта. Для снижения вероятности появления на свет ребенка с подобным диагнозом необходимо:
- вести здоровый образ жизни женщинам в период беременности;
- следить за адекватным протеканием гестации с регулярным посещением акушера-гинеколога;
- парам, решившим завести ребенка, пройти консультацию у генетика.
Несмотря на то что подобное заболевание невозможно излечить, благодаря соблюдению рекомендаций лечащего врача можно достичь благоприятного прогноза — снижения интенсивности проявления симптомов, при этом продолжительность жизни человека может составлять 50 лет.
Тем не менее не следует забывать про высокую вероятность внезапной смерти больного. У детей летальный исход наиболее часто наступает из-за наличия сопутствующих патологий внутренних органов и полиорганной недостаточности. У взрослых смерть зачастую развивается на фоне инсульта.
Осложнения и последствия
Главным осложнением у больных с синдромом Ретта является нейромоторные и ортопедические нарушения, ухудшение движений, невозможность самообслуживания и передвижения. Больные теряют способность целенаправленного движения и выполнения каких-либо действий.
Появление сколиоза может причинить боль в спине и пояснице, и в этом случае должен присутствовать массаж в лечении.
На первых стадиях ухудшается походка и в последующем больной совершенно перестает ходить, и прикован к инвалидному креслу. Одним из грозных осложнений является эпилептические припадки, которые становятся только чаще и длиннее по времени.
Причины возникновения синдрома
Причины возникновения продолжают изучаться. Особая роль отводится генетической предрасположенности, а также мутации в геноме, связанного с X хромосомой.
Мутация в гене, кодирующего метил-СрG-связывающий гликопротеин. приводит к нарушению правильного развития деятельности головного мозга в эмбриональном периоде развития.
Если ген не изменен, то мозг и его отделы развиваются без патологии. Если произошла мутация, происходит поражение X- хромосомы. Так как у женщин их две, то одна X-хромосома – нормальная, а вторая – дефектная.
С одной нормальной X-хромосомой девочкам удается появится на свет. Так как у мальчиков набор хромосом состоит из X и Y- хромосом, то их рождение с синдромом Ретта теоретически невозможна. Они погибают во внутриутробном периоде. Но существуют исключения.
При полисомии XXY хромосомах, рождение мальчиков возможно (синдром Клайнфельтера).
Симптомы
До 1 года никаких симптомов не наблюдается. Болезнь начинает проявлять себя в возрасте от 1 до 2 лет и только у девочек. Первые симптомы:
- Замедляется рост головы.
- Появляется отрешенность от окружающей действительности.
- Останавливается нормальное развитие психики, нарушается развитие функции узнавания и усвоения новой информации.
После этого прибавляются симптомы в двигательной сфере:
- Утрачивается способность брать и держать предметы рукой.
- Снижаются произвольные движения рук. Смена ведущей руки.
- Возникают специфические вращательные движения в кистях напоминающие протирание и мытье; битье себя по нижней челюсти; «заламывание» и заведение назад рук.
- Шаткая неуклюжая походка.
- Дрожь.
- Утрачивается или снижается речевая активность.
Также характерны дистрофические изменения кожных покровов: цвет кожи приобретает синюшный оттенок.
В дальнейшем отрешенность и нарушения движений могут снижаться. При достаточной психолого-воспитательной работе по развитию навыков могут незначительно восстанавливаться речь, способность к общению и уходу за собой.
Осложнения
Кроме развития умственной отсталости в виде сниженного интеллекта и неразвитых когнитивных функций развиваются грубые неврологические расстройства: эпилептические припадки, атрофия мышц спины с развитием искривления в позвоночнике, утрачивается способность ходить.
У детей
Болезнь развивается в раннем детском возрасте в 1 или 2 года. К окончанию детского и подросткового периода, к сожалению, формируются необратимые дефекты в психике и неврологическом состоянии. Из-за грубых неврологических нарушений до взрослого возраста доживают не все.
У девочек
Как было сказано выше, в связи с тем, что дефектный ген, вызывающий болезнь, располагается в X-хромосоме синдром Ретта наблюдается исключительно у девочек. Если эмбрион с наличием этого дефектного гена имеет мужской пол, то такой плод не развивается и погибает на ранних этапах беременности.
Почему синдром диагностируется только у девочек
Каждые 90 минут на свет появляется новорожденный с синдромом Ретта. Страдают этим заболеванием лишь девочки.
По доминантному типу наследования болеть будут преимущественно девочки, поскольку мутирует ген (MЕCР2), находящийся на плече X-хромосомы. У девочек таких хромосом 2. У мальчиков нет парной Х-хромосомы (у них XY), поэтому любая мутация гена, связанного с ней заканчивается летальным исходом среди младенцев мужского пола обычно еще во внутриутробном периоде, поскольку нет одной запасной здоровой хромосомы. Чаще причиной смерти становится перинатальная энцефалопатия. Однако продолжительность жизни девочек тоже сокращена до 30, максимум до 50 лет.
Известны единичные случаи, когда у мальчиков обнаруживали такую патологию, но они выживали. При наличии у лиц мужского пола полисомии – аномалии, характеризующейся дополнительной хромосомой, определяющей женский половой признак (синдром Клайнфельтера) возможно возникновение синдрома Ретта, где она из них будет дефектной, а вторая здоровой.
Диагностика
Синдром Ретта требует осуществления целого комплекса диагностических мероприятий. Прежде всего, детскому неврологу следует:
- ознакомиться с историей болезни пациента — для выявления сопутствующих патологий, которые могут усугубить течение недуга;
- собрать и проанализировать жизненный анамнез, в том числе изучить информацию о внутриутробном развитии плода и протекании беременности у женщин;
- провести тщательный неврологический осмотр — сюда стоит отнести оценивание двигательных и речевых навыков, походки и моторики. Помимо этого, требуется измерение окружности головы;
- детально опросить родителей больного — для установления степени выраженности симптоматики, что укажет на фазу и характер протекания болезни.
Наиболее информативными инструментальными обследованиями служат:
- МРТ головного мозга;
- электроэнцефалограмма;
- УЗИ внутренних органов.
Самой точной диагностической информацией обладают методики современной генетики, что дает возможность поставить правильный диагноз в пренатальном периоде, т. е. во время внутриутробного развития плода.
Причины развития и прогрессирования болезни Альцгеймера
Многие исследователи пытаются выяснить, почему с возрастом в тканях мозга человека начинаются дегенеративные процессы. Существует три гипотезы, объясняющие возможные причины развития данного заболевания:
- тау-гипотеза является одной из самых молодых, находящейся в стадии изучения. Согласно этой теории, патологические процессы в головном мозгу, приводящие к развитию деменции, запускаются при изменении структуры тау-белка. Сначала внутри нервных клеток образуются нейрофибриллярные клубки, вызывающие сбой в работе транспортной системы нейрона. Затем прекращается передача сигналов между отдельными клетками, после чего наступает и гибель нейронов;
- амилоидная гипотеза была открыта сравнительно недавно — ей еще нет 30 лет. В качестве основной причины развития дегенеративных процессов были названы скопления бета-амилоида. В поддержку этой теории было проведено много различных исследований, но вакцина, способная очистить мозг от белковых бляшек, также не является эффективной в ходе лечения деменции. Несмотря на то, что амилоидная теория научным сообществом принята в качестве основной, она не дает возможность объяснить все действия, происходящие в мозгу человека при развитии болезни Альцгеймера. Достоверно известно, что большое количество амилоидных бляшек не является непосредственной причиной заболевания — они лишь приводят к запуску дегенеративных операций. Что же действительно является пусковым механизмом к началу отложения бета-амилоида, пока не известно;
- холинергическая гипотеза была принята сразу после того, как началось изучение проблемы снижения умственных возможностей у пожилых людей. Ее авторы считали, что патология развивается в результате снижения синтеза ацетилхолина, являющегося нейромедиатором. С течением времени это предположение стало считаться маловероятным, так как медикаментозное снижение дефицита ацетилхолина малоэффективно при лечении деменции альцгеймеровского типа. Тем не менее, на основе этой теории было разработано большое количество методов поддерживающей терапии, многие из которых успешно используются и в нынешнее время.
Кроме того, многие исследователи считают, что к развитию болезни Альцгеймера может привести чрезмерное употребление сахара в пищу. Глюкоза блокирует выработку энзима, который необходим для предотвращения развития дегенеративных процессов в тканях головного мозга.
Что такое Синдром Ретта —
Прогрессирующее дегенеративное заболевание ЦНС предположительно генетического происхождения, встречается преимущественно у девочек, названо по имени австрийского ученого A. Rett, впервые описавшего его в 1966 г. Автор сообщил о 31 девочке с регрессией психического развития, аутистичным поведением, утратой целенаправленных движений и появлением особых стереотипных двигательных актов, «сжимания рук».
Распространенность
Частота его относительно высока — 1:10 000 девочек. В мире описано более 20 тыс. случаев заболевания; большинство из них спорадические, менее 1% — семейные. При изучении близнецов показана конкордантность по синдрому Ретта монозиготных и дискордантность дизиготных пар. Географическое распространение синдрома Ретта неравномерно. Отмечено скопление больных в определенных небольших сельских районах «Ретт-ареалы», что может быть связано с существующими популяционными изолятами. Такая концентрация заболевания наблюдается в Норвегии, Италии, Албании и Венгрии.
Что провоцирует / Причины Синдрома Ретта:
Подтверждена наследственная природа заболевания. Вопросы патогенеза заболевания остаются спорными. Генетическая природа связывается с ломкой Х-хромосомой и наличием мутаций в генах — регуляторах процесса репликации. Выявлены селективный дефицит ряда регулирующих рост дендритов белков, низкое количество глутаминовых рецепторов в базальных ганглиях, дофаминергических рецепторов в хвостатом ядре, нарушения холинергической функции. Гипотезу «прерванного развития», в основе которой лежит дефицит нейротрофических факторов, выдвинул D. Armstrong. Предполагается поражение нижних моторных нейронов, базальных ганглиев, вовлечение спинного мозга, ствола и гипоталамуса.
Анализ морфологических изменений при синдроме Ретта указывает на замедление развития мозга после рождения и остановку его роста к 4-летнему возрасту. Выявлено замедление роста тела и отдельных органов (сердца, печени, почек, селезенки).
Синдром Ретта у детей — что это такое и в чем опасность?
 Фото 1. Девочка с синдромом Ретта
Фото 1. Девочка с синдромом Ретта
Синдром Ретта – редко встречающееся генетическое заболевание, одним из главных признаков является поражение центральной нервной системы, которое влечет за собой потерю умственных и физических навыков, которые человек приобрел на протяжении своей жизни. Код МКБ-10 F84.2.
Так как это мутация конкретно Х-хромосомы, то это заболевание считается летальным для мальчиков, так как у них одна Х-хромосома. У девочек их две, поэтому угроза жизни не настолько велика. Синдром Ретта влечет за собой умственную недоразвитость и отставание в физическом развитии.
Впервые описал эту болезнь и выделил основные симптомы Андреас Ретт в 1960-1970 годы
Но особенно на эту хромосомную патологию обратили внимание группа ученых, которые под руководством Бенгта Хагберга сделали публикацию. Именно эти ученые и врачи описали симптомы деменции (развитие слабоумия), признаки аутизма, нарушения в мышечной системе и в движении рук у девочек
Исследования проводили сразу в трех странах Европы. Именно научная работа и наблюдение этих специалистов помогли медицине выделить синдром Ретта, как отдельное заболевание.